Hypofosfatazie plod. orogenic dysplazie
hypofosfatázie Je to vzácná autosomálně recesivní dědičná patologie, která se vyznačuje tím, kostní demineralizace a nízkou úrovní alkalické fosfatázy v séru a jiných tkáních. Působení alkalické fosfatázy v difosforečnanu a další komplexní fosfoefiry vede k akumulaci anorganického fosfátu, které jsou důležité pro tvorbu kostní trámčiny. Křehkost kostí v této nemoci je jen výsledkem jejich nedostatek rozvoje.
hypofosfatázie To je rozděleno do tří klinických typů, v závislosti na věku, kdy začíná nemoc: vrozené, novorozenců, dětí a dospělých hypofosfatázií. Vrozeně, neonatální, a ve většině případů, formy dětské mít autosomálně recesivní dědičnost, zatímco formulář pro dospělé - autosomální dominantnantny.
V vrozené neonatální variantou tohoto onemocnění se často vyskytuje smrt plodu nebo novorozenecké smrti.
Plodů s vrozenou hypofosfatázie mají všeobecný demineralizaci skeletu se zkrácením a zakřivení dlouhých kostí. Existuje několik zlomenin. To označilo demineralizace lebeční klenby, což vede k deformaci tvaru hlavy, pokud máte externí tlak na něj. Echografickém Tato funkce může být také v některých případech zjištěno, osteogenesis imperfecta typu II a ahondrogeneza 1A.
V literatuře existují náznaky prenatální diagnostika Toto onemocnění ultrazvukem a analyzuje úroveň alkalické fosfatázy v tkáňové kultuře choriových klků a amniotické buněk tekutin. Měření alkalické fosfatázy v plodové tekutiny sobě není spolehlivý způsob diagnostiky hypofosfatázií, protože hlavní část tohoto enzymu v plodové vodě způsobené provozem plodu gastrointestinálního traktu.

Enzymy, které nastane, když abnormalita hypofosfatázie, zastoupeny alkalické fosfatázy játra a kostní hmoty a způsobují pouze 16% celkové enzymatické aktivity plodové vody.
Nyní je možné provádět prenatální diagnostika děti typ hypofosfatázie pomocí molekulárně genetických studií choriových klků tkáně. Tam je popis ukázkového případu, kdy bylo zjištěno, že missense mutace při vyšetření u novorozenců s touto podmínkou.
Orogenic dysplazie je autosomálně recesivní choroba, charakterizované micromelia, koňská noha, mentálním postižením kartáče, více flexe kontraktury kloubů a skoliózou. Vzhledem k variabilitě fenotypových projevů její diagnostika při narození může být obtížné, a v případech mírné závažnosti diagnózy patologie je stanovena později.
klinické projevy rizomelicheskogotipa zahrnují zkrácení končetin, kontraktury, kartáče defekty s instalací palce v pozici únosu (palec „stopaři» - stopař palec) a těžkou abnormální instalace zastavení typu ‚jízda na nohy‘.
struktura hlava se obvykle nezmění, ale může být mikrognatie a rozštěp patra. Tento typ dysplazie doprovázena generalizovanou patologii chrupavkové tkáně s poruchou stavu jejího mezibuněčné látky a tvorby jizev pojivové tkáně a jejich následné osifikace. Jedná se o poslední proces vede k tvorbě kontraktur. Výsledky choroba mutací DTDST genu, což vede k narušené fosforylace proteoglykany.
 Vrozená idiopatická hyperkalcemie a hypofosfatázie. křehké kosti
Vrozená idiopatická hyperkalcemie a hypofosfatázie. křehké kosti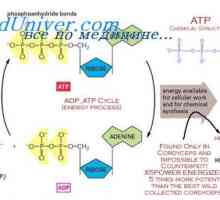 Multicystická ledvin dysplazie plod. Retsissivnaya fetální polycystické onemocnění ledvin
Multicystická ledvin dysplazie plod. Retsissivnaya fetální polycystické onemocnění ledvin Clinic plodů kosterní dysplazie. Diagnostika dysplazie kostní systému u plodu
Clinic plodů kosterní dysplazie. Diagnostika dysplazie kostní systému u plodu Posouzení plodu páteře zakřivení. Vyšetření vnitřních orgánů plodu
Posouzení plodu páteře zakřivení. Vyšetření vnitřních orgánů plodu Klasifikace ahondrogeneza plod. diagnóza ahondrogeneza
Klasifikace ahondrogeneza plod. diagnóza ahondrogeneza Kampomelicheskaya dysplazie plodu. Diagnóza kampomelicheskoy dysplazie
Kampomelicheskaya dysplazie plodu. Diagnóza kampomelicheskoy dysplazie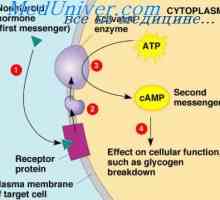 Osteogenesis imperfecta plod. Příčiny a diagnóza osteogenesis imperfecta
Osteogenesis imperfecta plod. Příčiny a diagnóza osteogenesis imperfecta Fibrohondrogenez a atelosteogenez. Ahondrogenez nebo anosteogenez
Fibrohondrogenez a atelosteogenez. Ahondrogenez nebo anosteogenez Vrozené zkrácení femuru. Štěpení rukou a nohou plodu
Vrozené zkrácení femuru. Štěpení rukou a nohou plodu Akromezomelicheskaya dysplazie. eykardi syndrom
Akromezomelicheskaya dysplazie. eykardi syndrom Genetické poruchy v ahondrogeneze. Achondroplazie v zárodku
Genetické poruchy v ahondrogeneze. Achondroplazie v zárodku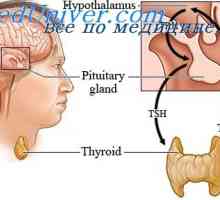 Hypofosfatazie. Hypoplastického levého srdce
Hypofosfatazie. Hypoplastického levého srdce Mezomelicheskaya dysplazie. Diagnózu a prognózu mezomelicheskoy dysplazie
Mezomelicheskaya dysplazie. Diagnózu a prognózu mezomelicheskoy dysplazie Osteogenesis imperfecta. Diagnostika a prognóza osteogenesis imperfecta u plodu
Osteogenesis imperfecta. Diagnostika a prognóza osteogenesis imperfecta u plodu Dominantní a recesivní alely chromozomů. dědičnost Autozomální dominanty
Dominantní a recesivní alely chromozomů. dědičnost Autozomální dominanty Klasifikace mukopolysacharidózy. Hurler nemoc, gentera, Sanfilippo, Morquio
Klasifikace mukopolysacharidózy. Hurler nemoc, gentera, Sanfilippo, Morquio Autosomálně recesivní polycystické onemocnění ledvin u dětí. Diagnostika a léčba
Autosomálně recesivní polycystické onemocnění ledvin u dětí. Diagnostika a léčba Shvahmana-Diamond syndrom. Metafizariaya typ chondrodysplasia makové-Cusick a ukládání glykogenu
Shvahmana-Diamond syndrom. Metafizariaya typ chondrodysplasia makové-Cusick a ukládání glykogenu Kmen předloktí
Kmen předloktí Caroli choroba
Caroli choroba Jiné patologie novorozenců
Jiné patologie novorozenců