Klasifikace mukopolysacharidózy. Hurler nemoc, gentera, Sanfilippo, Morquio
Typ Mukopolysacharidosa I (Hurler nemoc) -nasledovanie autosomálně recesivní. Buňky a intercelulární látka uložena dermatapsulfat, heparansulfát a gangliosidy. Tam je nedostatek enzymu beta-galaktosidázy, a poslednimdannym a-iduronidazy.
Tam byly závažné příznaky gargoilizma s progresivními duševních poruch, předčasného zakalení rohovky, hluchoty a hepato-splenomegalie. V krevních lymfocytů s granulí s metachromatické v moči se prudce zvýšil obsah mukopolysacharidů. Děti umírají ve věku 10-12 let s příznaky poškození mozku (hydrocefalus), srdce nebo ze interkurentních infekcí.
Typ Mykopolisaharidoz II (Gentera choroba) -nasledovanie recesivní, spojené s X-hromosomon, která je uložena v tkáních dermatansulfátů a heparansulfátů, na nedostatky | 3-galaktosidázy a podle posledních údajů, nedostatek enzymu a-sulfatázy iduronosulfat [Swift Th, Mac Donald Th, 1976 .. ]. Onemocnění provozuje dlouho ve srovnání s Hurler onemocnění, symptomy nejsou tak výrazné, že není žádný zákal rohovky. V moči, množství mukopolysacharidů zvýšila, v krevních lymfocytů - granule s Metachromatická.
Typ Mykopolisaharidoz III (Sanfilippo nemoc) - dědičnost autosomální recesivní. V tkáních nahromaděné heparansulfátu. Tam je nedostatek beta-galaktosidázy. Kromě toho, snížená aktivita v orgánech geparansulfatazy [Cain H. et al., 1976]. Výše somatických změn převládá psychomotorickou retardací. Nicméně, podle N. Cain et al. (1976), obsah GAG v játrech zvýšená 12,5 krát proti standardům, především v důsledku hromadění heparansulfátu. Podobná látka se nachází v neuronech v mozku, v tepně stěn, sleziny. Množství GAG moči zvyšuje, v krevních lymfocytů - Metachromatická granulí.
Mukopolysacharidóza typ IV (Morquio nemoc) -nasledovanie autosomálně recesivní. V tkáních kerataisulfat zpoždění vzhledem k N-atsetilgeksozaminsulfatazy deficitu. Převládající kosterní změny a zákalu rohovky. Granule v lymfocytech chybí.
Typ V Mykopolisaharidoz (Sheye nemoc) -to v současné době považován za variantu mukopolysacharidózy typu I, se klinicky projevuje u dospělých, od aktivity enzymu a, L-iduronidazy mírně snížena.
Typ mukopolysacharidóza VI (Maroto onemocnění - Lamy) - dědičnost je autozomálně recesivní.
V tkáních je zpožděn dermatan. Tam je nedostatek enzymu 4-acetylgalaktosamin-4-sulfatázy. Zbytek je podobná typu mukopolysacharidóza onemocnění I, avšak porušení psychomotorického vývoje je velmi nízká nebo chybí.
Typ Mukopolysacharidóza VII vyznačující se deficiencí enzymu beta-glukuronidázy. Tkáň se akumuluje a je vylučován do dermatan moči. Projevy nemoci jsou podobné mukopolysacharidóza typu I, ale ne tak výrazný.
mukopolysacharidóza typicky diagnostikovány klinické příznaky, a také na základě biochemické stanovení GAG v moči i přítomnost Metachromatická granulí do krve lymfocytů. Histochemické studie obtížné, protože GAG velmi snadno rozpuštěných při fixaci tkáně, a morfologické změny maloharakterny. Pokud dojde k onemocnění v rodinách, doporučuje se k amniocentéza, studii amniové kultury a amnion buněk tekutiny na přítomnost GAG.
Takže na závěr, je třeba poukázat na to, že i přes převládající směnné porušování GAG v takzvaných „čistých“ mukopolysacharidových poruch, které jsou vlastně skupina onemocnění, při kterých jsou ve většině případů porušování smíšených typově mukopolysacharidů a gangliosidy.

 Vrozená idiopatická hyperkalcemie a hypofosfatázie. křehké kosti
Vrozená idiopatická hyperkalcemie a hypofosfatázie. křehké kosti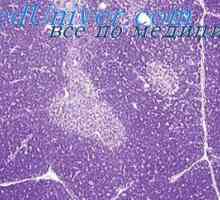 V kombinaci s imunodeficience fermentopathy. nezelofa syndrom nebo alymphocytosis
V kombinaci s imunodeficience fermentopathy. nezelofa syndrom nebo alymphocytosis Hypofosfatazie plod. orogenic dysplazie
Hypofosfatazie plod. orogenic dysplazie Autozomálně recesivní dědičnost. dědičnost X-vázaná
Autozomálně recesivní dědičnost. dědičnost X-vázaná Glyukotserebrozidoz Gaucherova choroba. Glikosfingolipidoz Fabryho choroba
Glyukotserebrozidoz Gaucherova choroba. Glikosfingolipidoz Fabryho choroba Gangliosidóza. Sandhofa onemocnění a juvenilní gangliosidóza
Gangliosidóza. Sandhofa onemocnění a juvenilní gangliosidóza Mucolipidosis: mannozidoz a fucosidosis. Juvenilní sulfatidoz typ oustina
Mucolipidosis: mannozidoz a fucosidosis. Juvenilní sulfatidoz typ oustina Galaktoziltseramidoz Krabbe onemocnění. Sulfatidoz metachromatická leykodistrosriya Scholz
Galaktoziltseramidoz Krabbe onemocnění. Sulfatidoz metachromatická leykodistrosriya Scholz Amavroticheskaya vrozená hloupost. Mucolipidosis
Amavroticheskaya vrozená hloupost. Mucolipidosis A leukodystrofií mukopolysacharidózy. Riziko mukopolysacharidosy
A leukodystrofií mukopolysacharidózy. Riziko mukopolysacharidosy Pompeho choroba Pompeho nemoc. Klinika a diagnostika Pompeho nemoci
Pompeho choroba Pompeho nemoc. Klinika a diagnostika Pompeho nemoci Glykogen spalničky skladování choroba, Andersen McArdl. Hersey nemoc, Thomson, kontejnery
Glykogen spalničky skladování choroba, Andersen McArdl. Hersey nemoc, Thomson, kontejnery Retikulární dysgeneze. purinová nukleosidová fosforyláza deficit
Retikulární dysgeneze. purinová nukleosidová fosforyláza deficit Transplantace kmenových buněk pro akumulaci onemocnění a talasemie
Transplantace kmenových buněk pro akumulaci onemocnění a talasemie Vimizim schválen k léčbě syndromu Morquio
Vimizim schválen k léčbě syndromu Morquio- Výborná lékařská encyklopedie IC nevronet. choroba
 Porušení lipidového metabolismu
Porušení lipidového metabolismu Peroxisomovým nemoc
Peroxisomovým nemoc Neyrometabolicheskie nemoc
Neyrometabolicheskie nemoc Mucolipidosis
Mucolipidosis Lysosomální onemocnění u dětí
Lysosomální onemocnění u dětí