Biochemické prenatální diagnóza cystické fibrózy, adrenální hyperplazie.
Od roku 1980 do roku 1985, tj. E. Před výskytem molekulárních technik, prenatální diagnóza cystické fibrózy To provádí pouze na základě prostudování aktivitu určitých enzymů ve střevech plodu AF. PD idea této nemoci tím, že studuje činnost enzymů střevní mikroklcích vznikl vzhledem k tomu, že velká část se skládá z epitelu plodových buněk mezi buňkami AF.
Bylo zjištěno, že Charakteristické pro plod pacienta změny spektra proteinů AF stanovených v relativně krátkou dobu (16 až 20 týdnů těhotenství), a zřejmě jsou výsledkem poruch střevní propustnosti vzhledem k přechodné nebo persistiruyushego ileus. Britská výzkumná skupina byly vyvinuty a hlavní kritéria pro ověřování diagnózu cystické fibrózy u plodu, jako mekonia a Leus, vyvýšená albuminu v smolky tenkém střevě, přítomnost změn v vývodem pankreatu.
I přes rozvoj molekulární genetiky Studie o cystické fibrózy, Diagnóza tohoto onemocnění pomocí molekulárních metod je často významně brání vzhledem k tomu, že ve vysoce rizikových rodinách, kde pacient s cystickou fibrózou dítě zemřel na cystické fibrózy genové mutace nelze určit. V důsledku toho se stává nemožným přímých i nepřímých molekulární diagnostiku. Velmi často taková situace nastane v naší zemi, kde molekulární metody identifikované ne více než 65-70% z mutovaných chromozomů, a počet vysoce rizikových rodin, hledají PD cystickou fibrózou, kteří mají nemocné dítě zemřelo, a to až do výše 80%. Všechny tyto rodiny budou považovány za zcela uninformative, tj nevhodné pro PD molekulárních metod. Pokud byla genová mutace transmembránový protein identifikován pouze jednoho z rodičů a studie polymorfismu v nepřítomnosti nemocného dítěte, je bezvýznamné, rodina se považuje za částečně informativní. Biochemické PD cystická fibróza v trimestru II se koná v částečně informativních rodin, pokud je plod bylo známo mutaci od jednoho rodiče, nebo zcela neinformativní rodin.

V naší zemi, Biochemické prenatální diagnóza cystické fibrózy To bylo vypuštěno v roce 1986 v Ústavu experimentální medicíny, Akademie lékařských věd SSSR (St. Petersburg), a v roce 1987 se konala v vytvořený na Ústavu gynekologie a porodnictví. DO Otga unie a nyní ruský centrum prenatální diagnostiky cystické fibrózy. Technika biochemické diagnostiky cystické fibrózy je uveden v revizi.
Biochemické prenatální diagnóza kongenitální adrenální hyperplazie
Jedním z nejvíce společný fermentopathia Je kongenitální adrenální hyperplazie (CAH), způsobené porušením syntézy kortizolu v důsledku nedostatku enzymu 21-hydroxylovaný. Výskyt tohoto onemocnění u novorozenců je 1/5000 - 1/15 000 Onemocnění je autosomálně recesivní dědičnost a vyznačuje se klinicky významné a genetických polymorfismů.
PD solteryayuschey formy adrenální hyperplazie ve čtvrtletí II metody těhotenský hormon z výzkumu prováděného od poloviny 70. let. V Rusku jsou tyto výzkumy byly prováděny v Moskvě NTsAGiP RAMS. Amniocentéza se provádí při 9-12 nebo 16-24 týdnech těhotenství. Uvažovaná 10-11 týdnů optimální doba, kdy plod při identifikaci pacienta může ukončit těhotenství do 12 týdnů. Diagnóza se provádí stanovení hladiny AF-17 oksiprogesterona metodou radioimunoanalýzu. Nicméně je třeba připomenout, že v současné době více než je považován za bezpečný a efektivní způsob diagnostiky DNA adrenogeni-tal syndrom.
21-hydroxylázy gen identifikovány a mapovány. Studoval v detailu nejčastější hlavní mutace charakteristické pro ruského lidu. Molekulární diagnostika CAH se koná v lékařské genetiky Research Center (Moskva) a Institutu porodnictví a gynekologii. DO Ott RAMS (Sankg- Petersburg).
 Anomálie slinivky břišní
Anomálie slinivky břišní Cirhóza slinivky břišní
Cirhóza slinivky břišní Polycystických, cystická fibróza (mukoviscidóza) pankreas
Polycystických, cystická fibróza (mukoviscidóza) pankreas Příčiny a mechanismy vývoje teratomů krku. Diagnóza plodu krk teratomů
Příčiny a mechanismy vývoje teratomů krku. Diagnóza plodu krk teratomů Druhy cystický hygrom plod. Diagnóza cystické hygromy
Druhy cystický hygrom plod. Diagnóza cystické hygromy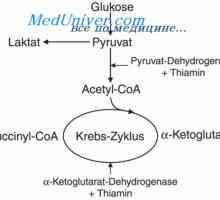 Uzi stav břicho. Mekonium peritonitidy u plodu
Uzi stav břicho. Mekonium peritonitidy u plodu Diagnostika smolky neprůchodnosti střev. Tenkého střeva plodu s cystickou fibrózou
Diagnostika smolky neprůchodnosti střev. Tenkého střeva plodu s cystickou fibrózou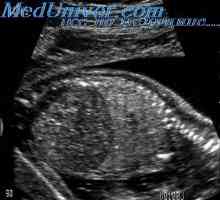 Plodu slinivka Uzi. Vyšetření žlučníku plodu
Plodu slinivka Uzi. Vyšetření žlučníku plodu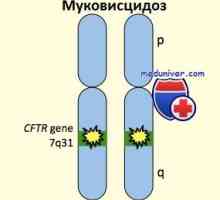 Žloutenka u dítěte s cystickou fibrózou
Žloutenka u dítěte s cystickou fibrózou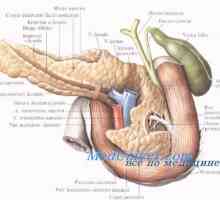 Metody hodnocení exokrinní slinivky
Metody hodnocení exokrinní slinivky Zjištěno protein, který způsobuje renální fibrózy
Zjištěno protein, který způsobuje renální fibrózy- Diagnostika prodloužené těhotenství
 Načasování prenatální diagnostiku genetických chorob.
Načasování prenatální diagnostiku genetických chorob. Kombinovaná gen diagnostika onemocnění. Biochemické metody v prenatální diagnostiku.
Kombinovaná gen diagnostika onemocnění. Biochemické metody v prenatální diagnostiku.- Cystická fibróza (cystická fibróza slinivky břišní). Etiologie, patogeneze. Těžký dědičné…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
 Cystické nádory pankreatu
Cystické nádory pankreatu Cystická fibróza: symptomy, léčba, diagnostika, příznaky, příčiny
Cystická fibróza: symptomy, léčba, diagnostika, příznaky, příčiny Enteropatie novorozenci, příznaky, léčba
Enteropatie novorozenci, příznaky, léčba Cystická fibróza je dítě, příčiny, příznaky, léčba
Cystická fibróza je dítě, příčiny, příznaky, léčba Mekonium ileus, korek
Mekonium ileus, korek