Žloutenka u dítěte s cystickou fibrózou

Cystická fibróza - porušení elektrolytů dopravy v epiteliálních buňkách. Část této patologie může být diagnostikována zvýšenými hladinami chloridu v potu kapaliny. Vzhledem k tomu, abnormální elektrolyt transport přes epitel je základem patologických změn u cystické fibrózy, byly provedeny identifikaci a klonování genu v části chromozomu 7 kódování CFTR.
prokázaly, že CFTR Je to nejen substrátem pro aktivaci fosforylace chloridový kanál, ale ve skutečnosti kanálem chloridu regulovaného cAMP. Bylo zjištěno, že cystická fibróza je spojen s asi 1000 CFTR genové mutace, z nichž nejběžnější je F508 (trojsytného vypuštění, nahrazení fenylalaninu v 508 th poloze).
Ve Spojených státech, mutace F508 ve více než 90% pacientů s cystickou fibrózou. Mechanismus, kterým CFTR mutace vyvolat onemocnění zahrnuje redukci (nebo nepřítomnost) syntézy CFTR, zrání defektu protein předčasnému rozpadu, dysregulace funkce CFTR, defektu vodivosti chloridů nebo zvýšené zničení CFTR.
ačkoli cystická fibróza lépe známý narušit sekreci chloridu v potních žláz a dýchacích defektních CFTR forem jsou rovněž zodpovědné za zahuštění pankreatické šťávy a tajemství hepatobiliárním systémem. Porážka hepatobiliárního systému je nejčastější příčinou úmrtí v mimoplicních cystickou fibrózou.
novorozenci choroba mohou mít akutní účinky ve formě cholestatické žloutenky, a to zejména v případech, kdy bylo dítě slaveny mekonia ileus. Normální CFTR apikální zóna se nachází podél cytoplazmatických membránách epitelových buněk ve žlučových routes- hepatocytech ne. Apikální lokalizace CFTR v buňkách žlučových cest pomáhá pochopit jak chlorid kanál CFTR nastavitelný, se podílí na normální sekreci žluče.
Video: rozprašovač terapie cystické fibrózy
Klasické projevy cystická fibróza patří malabsorpce u kojenců ve spojení se zvýšeným výskytem infekcí dýchacích cest v dětství a dospívání. Mnoho novorozenecké onemocnění se projevuje střevní obstrukce kvůli ileus meconium. Jen málo dětí Choroba se projevuje na klinice neonatální cholestázou, připomínající žlučových atrézie.
U starších dětí, někdy diagnóza "cirhóza„Nebo“portální hypertenze„Předchází zřízení“ cystické fibrózy „diagnózy. Mnoho pacientů s těžkým poškozením jater kvůli poruchám průtoku žluči a následného rozvoje žlučových transplantaci cirhóza jater je nutná. Kromě chirurgického zákroku (to znamená, transplantace jater) jediným možným konzervativní terapie je léčebná metoda kyseliny ursodeoxycholové, když je působení léčiva v progresi onemocnění jater, je nejasný.
S objevem skutečnosti, že CFTR lokalizovány v prostoru se nachází v submukózních žlázách a povrchu epitelu, tyto lokusy mohou být důležité, pokud jde o budoucí vývoj metod pro cílenou genovou terapii. Tak, očekává se, že může být zabráněno respirační příznaky cystické fibrózy, nebo převod exprese normálního CFTR cDNA k epiteliálním buňkám intrahepatálních žlučovodů. Tyto žlučovodu buňky mohou být zkoumány za použití adenoviru jako vektoru vadného CFTR.
Tato potenciálně preventivní léčby jaterních onemocnění způsobených cystická fibróza, s pomocí geneticky CFTR v žlučovody také dává příležitost prozkoumat a vytvořit mechanismy odpovědné za vznik hepatobiliární systém patologie u dětí s cystickou fibrózou.

 Polycystických, cystická fibróza (mukoviscidóza) pankreas
Polycystických, cystická fibróza (mukoviscidóza) pankreas Diagnostika smolky neprůchodnosti střev. Tenkého střeva plodu s cystickou fibrózou
Diagnostika smolky neprůchodnosti střev. Tenkého střeva plodu s cystickou fibrózou Metafiznaya chondrodysplasia Jensona. Poruchy syntézy kolagenu v plodu
Metafiznaya chondrodysplasia Jensona. Poruchy syntézy kolagenu v plodu Abnormality SOx genů a TVH Holt-Oram syndromu. Fibroblastové růstové faktory
Abnormality SOx genů a TVH Holt-Oram syndromu. Fibroblastové růstové faktory Předčasná puberta je spojena s genetickými mutacemi, dědičná otcovská
Předčasná puberta je spojena s genetickými mutacemi, dědičná otcovská Vliv antibiotik na střevní antimikrobiálních peptidů
Vliv antibiotik na střevní antimikrobiálních peptidů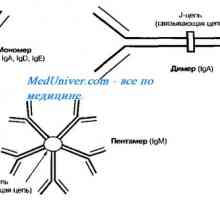 Autozomálně recesivní syndrom hyperproduction imunoglobulinu m (IgM). genová mutace pomoc
Autozomálně recesivní syndrom hyperproduction imunoglobulinu m (IgM). genová mutace pomoc Mechanismy rozpad a katabolismus proteinů v organismu
Mechanismy rozpad a katabolismus proteinů v organismu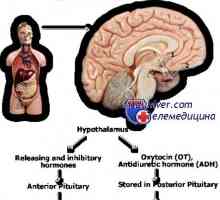 Mutace gonadotropin geny. Mutace v podjednotky LH a FSH
Mutace gonadotropin geny. Mutace v podjednotky LH a FSH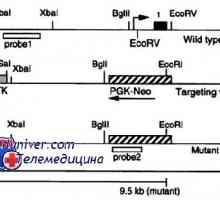 Mutace gonadotropin receptory. Abnormality LH a FSH receptoru
Mutace gonadotropin receptory. Abnormality LH a FSH receptoru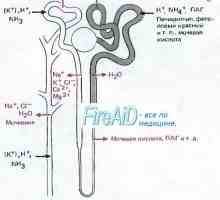 Gitelmanova syndrom u dětí. Diagnostika a léčba
Gitelmanova syndrom u dětí. Diagnostika a léčba- Cystická fibróza (cystická fibróza), příčiny a příznaky, léčba a komplikace cystické fibrózy
- Nalezeno léčení vrozených onemocnění očí
- Jaké jsou genetické choroby
 Genetické poruchy, které vedou k neplodnosti u lidí
Genetické poruchy, které vedou k neplodnosti u lidí Biochemické prenatální diagnóza cystické fibrózy, adrenální hyperplazie.
Biochemické prenatální diagnóza cystické fibrózy, adrenální hyperplazie.- Genetické choroby jsou jedny z nejčastějších onemocnění
- Nová kombinace léčiv prodloužit životy lidí s cystickou fibrózou
 Cystická fibróza: symptomy, léčba, diagnostika, příznaky, příčiny
Cystická fibróza: symptomy, léčba, diagnostika, příznaky, příčiny Nemoci metabolitů dopravu
Nemoci metabolitů dopravu Cystická fibróza je dítě, příčiny, příznaky, léčba
Cystická fibróza je dítě, příčiny, příznaky, léčba