Xy karyotypu 46 mužů

Karyotyp 46, XY.
Čistý gonadální dysgeneze (Suayra syndrom)
Existují dvě teorie o patogenezi tohoto onemocnění. Za normálních okolností jsou zárodečné buňky začnou mít vliv na vývoj pohlavních žláz až po přestěhování do sexuálních šňůry. Předpokládá se, že u některých pacientů s čistým gonadální dysgenezí nedochází tento pohyb, a místo toho, varlat vytvořily tyazhevidnye pohlavní žlázy. Jiní pacienti vykazují mutace genů zodpovědných za vývoj varlat, což vede v zárodečných buňkách nepronikají do mozkové substance indiferentní gonády zůstávají v kortikálních a rychle umírají. V přítomnosti Y chromozomů reprodukčních buněk usmrtí úplně. V 15% pacientů s čistými pohlavních SRY genových mutací dysgeneze zjištěno, zbytek, zdá se, že existují i jiné mutace genů podílejících se na testikulárního vývoje. Popsaný X oky forma onemocnění je samozřejmě vzhledem k tomu, že geny jsou umístěna pod SRY, jsou rozhodující při diferenciaci pohlavních žláz. U těchto pacientů je normální vývoj Mullerian a vytvoří samice fenotyp.
U pacientů s čistým gonadální dysgenezí 46, XY samice fenotyp charakteristika a zpoždění sexuálního vývoje. Naopak u pacientů s Turnerovým syndromem, které mají poměrně vysoký růst, protože za prvé, žádné růstové léčba epifýzy zóny pro dlouhou dobu zůstat otevřená vzhledem k nedostatku pohlavních hormonů, a za druhé, ve směru Y-chromozomu jsou geny, které urychlují růst , Hladiny hormonu uvolňujícího gonadotropin v krvi výrazně zvýšil. V laboratorní studie ukazují karyotypem 46, XY. Riziko maligní transformace tyazhevidnyh pohlavních žen s onemocněním je 25 až 35% - vyšší než u jiných forem gonadální dysgenezí s Y-chromozomu. Jakmile je diagnóza je nezbytná k odstranění pohlavních žláz. Někdy tumory zárodečných buněk v pubertě začnou vylučovat testosteronu nebo estrogenu, což vede k různým stupněm feminizaci nebo virilization.
Přetrvávání Mullerian
V normální sexuální diferenciace mužského migraci a diferenciaci zárodečných buněk varlat vzorek začíná ihned po aktivaci genů nimi genově řízené. Později začíná syntetizovaný antimiillerický hormon, má místní inhibiční účinek na rozvoj jejich derivátů. Müllerian přetrvávání může být způsobena mutací genu antimyullerova hormonů (jako důsledek úrovně faktoru v krvi je velmi nízká, nebo není detekován), nebo defekt receptoru proteinu. Výsledkem je, že u pacientů s vnějším fenotypem jsou také děloha a vejcovody. V nepřítomnosti jiných genetických poruch varlat vyvíjejí normálně a produkovat dostatečné množství androgenů.
U pacientů s přetrvávající Mullerian z důvodu porušení procesu sestupu varlat se obvykle vyskytuje jednou nebo bilaterální kryptorchizmu a tříselná kýla. Někdy se odhalí vady chámovodu. Odstranění dělohy a pochvy je spojena s vysokým rizikem poškození chámovodu a následné neplodnosti.
anorchidism
Při anorchidism varlata začnou vyvíjet se normálně a vyrábět antimiillerického hormonu, avšak před syntézu testosteronu podstoupit atrofii. Dříve se za to, že důvodem jsou vaskulární poruchy nebo změny v okolní tkáni, avšak vzhledem k dvoustranné léze, zdá se pravděpodobné narušení na molekulární úrovni. Po regresní Müllerian syntézy testosteronu a diferenciace volfovyh potrubí nedochází.
Fenotyp anorchidism jsou různé a závisí na fázi pohlavního vývoje, k němuž došlo atrofii varlat. Je pravidlem, že u pacientů se rodí s ženským fenotypem a rodiče přivést k lékaři, protože se nevyskytují puberty. V laboratorní studii ukazují karyotyp 46, XY a vysoké úrovně gonadotropních hormonů. Když audit pánev nezjistí pohlavních žláz zvuk nebo derivátů pohlavních cest.
Porušení syntézy a působení androgenů
U pacientů s karyotypem 46, XY, a porušení působení androgenů differeniirovka varlata obecně pokračuje normálně a antimyullerova sekreci hormonu netrpí. Příčiny syntézy a působení androgenů:
- Syntéza a akce LH poruchy;
- Nedostatek enzymů zapojených do syntézy androgenů;
- vady na androgenní receptor.
Porušení syntézy a působení LH
Leydigovy buňky jsou umístěny na receptoru Ai vázání těchto receptorů k ligandu spouští syntézu androgenů ve varlatech. stimulace LH receptor v důsledku vysokých hladin hCG v séru matky probíhá před zahájením sekrece LH hypofýzou plodu. Pacienti s 46.XY karyotypem detekován genové mutace receptoru GnRH a LH podjednotky genu. Během těhotenství je hladina hCG je dostatečně vysoká, aby spustit syntézu testosteronu ve varlatech. Při narození, dítě má normální mužský fenotyp, takže diagnóza je vyrobena pouze v období dospívání, po vyhledání lékařské pomoci v důsledku zpoždění puberty. LH genu receptoru mutace u pacientů s karyotypem 46, XY je pravděpodobné, že naruší tvorbu mužského fenotypu, protože hCG se neváže na LH receptorům a není schopen stimulovat syntézu androgenu v plodu. Příznaky onemocnění se podobají odolnost vůči androgenů.
Porušení syntézy androgenů
Při selhání nebo nepřítomnosti enzymů zapojených do syntézy androgenu, androgen sekrece je snížena, a vývoj pohlavních cest a vnější genitálie rušen. Syntéza androgenů mohou být rozděleny na různých stupních, a stupeň selhání určitého enzymu se může lišit - od menšího snížení aktivity až do jeho úplné nepřítomnosti. Klinický obraz je rozmanitá: existují jednostranné nebo častěji bilaterální kryptorchismus, mikropěnu. Všechny nemoci se dědí autosomnoretsessivno.
Kongenitální adrenální hyperplazie lipondnaya
Toto vzácné onemocnění je způsobeno geneticky podmíněných poruch steroidogeneze v rané fázi. Akutní reakce reguluje dodání steroidů cholesterolu z vnější do vnitřní mitochondriální membrány a spoušť tohoto procesu je protein hvězda. Permanentní steroidů kapacita CYP11A1 transkripce genu je určena. Genové mutace STAR porušuje ne všechny steroidogenez- malý podíl hvězdicovité nezávislé steroidogeneze ztracené později v souvislosti s poškozením sekundární buňky, což vysvětluje možnost pozdě, několik měsíců po narození, příznaky nedostatečnosti nadledvin. Podobný syndrom je vzácnou příčinou mutace CYP11A (nyní popsány dvě varianty) kódující P450ssc.
3-hydroxysteroid dehydrogenázy
3 -gidroksisteroidtsegidrogenazy nedostatek vede ke zvýšení hladiny dehydroepiandrosteronu a nižší hladiny pohlavních hormonů - testosteronu a androstendion. Pro onemocnění charakterizované ztrátou syndrom soli, hyponatremie a hyperkalemie znamení. Pokud se doba nedělá diagnózu a zahájit léčbu, novorozenec se může vyvinout nedostatečnost nadledvin. -gidroksisteroidtse enzym 3-hydrogenase je kódován dvěma geny - HSD3B1 a HSD3B2. U pacientů s deficitem enzymu odhalí mutace HSD3B2 gen.
Hydroxyláza deficit 17h
deficit syndrom 17a-hydroxylázy - vzácná genetická porucha steroidní biosyntézy, což má za následek snížení produkce glukokortikoidů a syntézu pohlavního steroidu a zároveň zvýšit mineralokortikoidní prekurzory. Cytochrom P450c17 považovány bifunkční enzym, kteří mají obě 17 a -gidroksilaznoy 17,20-lyázy aktivitu a kódovaný CYP17 gen. Nedostatek různých enzymových funkcí v důsledku různých mutací v genu. P450c17 je detekován v paprsku a oblast ok v nadpochechnikov- kůra zona glomerulosa odpovídající steroidní biosyntézu žádné větve, takže mineralokortikoidní syntéza není narušen. Čistý steroidogeneze zóna, naopak, je rozdělen v rané fázi a produkce androgenu neprobíhá, v důsledku čehož, vnější genitálie, bez ohledu na genetické pohlaví ženy. Může vyvinout hypokalémie a hypertenzi v důsledku nahromadění prekurzoru steroidních hormonů a formace nadbytku mineralokortikoidů (např deoxykortikosteronu). Hladiny ACTH, FSH a LH v krevní plasmě u těchto pacientů je obvykle zvýšená. U pacientů s karyotypem 46, XY děloha a vaječníky ne vizualizovat. Léčba glukokortikoidů a draslík-šetřící diuretika přispívá k normalizaci krevního tlaku a prevence ztráty draslíku.
$ 17-hydroxysteroid dehydrogenázy
17 selhání-hydroxysteroid-idrogenazy peněz vede k narušení přeměnou androstendionu na testosteron je charakterizována akumulací dehydroepiandrosteron a androstendionu a snižují syntézu testosteronu, a, nakonec, dihydrotestosteron. Bez léčby, přebytek androstendionu se převede na estron, který stimuluje vývoj mléčné žlázy. Zjistili jsme, Několik mutací -gidroksisteroiddegidro genu 17-dehydrogenázu.
Nedostatek 5a-reduktázy
V normální sexuální diferenciace Leydigovy buňky produkují testosteron, který je v cílových buňkách působením 5-reduktázy převede na účinnější androgen, dihydrotestosteron. Testosteron a dihydrotestosteron působí na stejné receptory androgenů dihydrotestosteronu, ale menší disociační konstanty, přičemž se váže na receptor silnější a má silnější účinek. Na 5 úrovní reduktázy nedostatek testosteronu normální nebo zvýšené. Tato choroba vede ke konzistentním porušování vnějších genitálií. Testosteron poskytuje normální vývoj volfovyh potrubí, protože 5 reduktázy není zapojen do tohoto procesu.
Před pubertou rozlišit selhání 5a-reduktázy syndromem androgenní odolnosti velmi obtížné, a to zejména v případě, že vejce jsou odstraněny před nástupem puberty. Uložíte-li varlata v průběhu dospívání se často vyskytuje u pacientů virilization. Stejně jako v normálním pubertě, v tomto období je prudký nárůst ve varlatech syntézy testosteronu. Výsledkem je, že i přes pokračující nedostatek 5a-reduktázy, je množství testosteronu, je dostatečné k poskytnutí lokálního účinku v cílových buňkách. Vnější genitálie se začínají transformovat samce tipu- možné vytvořit plnohodnotnou penis. Tento jev byl pozorován u velkého rodinného studia v Dominikánské republice, kde byli tito pacienti s názvem guevedoces, což znamená, že varlata do 12 let. Ukládání ženského fenotypu u dospělých s deficitem 5a-reduktázy dochází poměrně často.
Enzym 5 reduktáza je kódován dvěma geny - srd5a1 a SRD5A2. SRD5A2 gen je exprimován v oblasti genitálií, takže bylo jeho mutace vedou ke klinickému obrazu je popsáno výše. SRD5A1 gen je exprimován v kůži, zejména pokožku hlavy.
Vady androgenní receptor
Důsledky rezistence na androgenu dochází na fázi zevního genitálu. U těchto pacientů, Müllerian atrofovaly varlata vyvíjejí normálně a vylučují dostatečné množství testosteronu, který v periferních tkáních je přeměněn na dihydrotestosteron. Nicméně, androgenní receptory jsou chybí nebo není schopen vázat se na androgen-mi, takže normální samčí fenotyp nevyvíjejí.
Struktura a androgen gen receptoru mutace studované docela dobře. Je umístěn na dlouhém rameni chromozomu X a obsahuje 8 exonů v každém z nich může být mutace (většinou bod) a vedou k různým formám syndromu androgenní odporu - mírné dokončit. Oba plné a částečné odolnost proti androgen dědí recesivní, spojka s X-chromozómu. V rodinách různých pacientů ukazují zjištěna extrémně vzácné unikátní mutatsii- stejná mutace, takže vyvinout metodu pro identifikaci genetického odolnost proti androgenů vadou jediného genu nemůže být. Různé mutace vedou k nerovnoměrné receptory vad. Nejčastěji se porušila nukleotidovou sekvenci kódující gormonsvyazyvayuschy doménu. Jiné mutace ovlivňují DNA vazebnou doménu. V tomto případě je funkce receptoru androgenu netrpí, ale komplex hormon - receptor je schopen komunikovat s specifickým místům DNA (postreceptor vady). defekty Míra receptoru se liší od úplné androgenní necitlivosti na téměř normální hormonální přenos signálu.
Kompletní odpor androgeny (testikulární feminizace)
Normálně vyvinuté varlata, uspořádané v dutině břišní, ingvinální kanál nebo stydké pysky, na spermatogenezi, ale chybí nebo je neúplná. Po dokončení sexuální zrání vajec, které mají být odstraněny, protože riziko jejich maligní transformace je 2-22%. Někdy otok dochází před nástupem puberty však brzy odstranění varlat produkují nedoporučuje, protože jsou důležité pro vývoj ženských charakteristik sekundárních pohlavních a psychosociální další úpravy. Blokuje působení androgenů v režimu offline, takže prsní žlázy může dosáhnout velké razmerov-, vyznačující se tím, že se skládá převážně z tukové tkáně.
Dílčí odpor na androgeny (částečné androgenní necitlivosti syndrom)
Jedná se o vzácné onemocnění než testikulární feminizace. Klinické projevy syndromu měnit. Pacienti jsou obvykle zvýšen jako dívky. V dospívání, pubické ochlupení se zvyšuje a zvyšuje klitoris.
Fenotyp pacientů může být muž nebo žena, s maskulinizace vyjádřený v různé míře. Někteří pacienti našli hypospadias, jiní k nerozeznání od zdravých mužů a trpí jen gynekomastie a neplodnost.
Není endokrinní příčiny vývojové poruchy mužských pohlavních orgánů
Během běžné sexuální diferenciace mužského typu lze také pozorovat poruchy sexuálního vývoje. Obvykle se izolované vady: nedostatek varlat nebo penisu, penis dysplazie (zdvojnásobení epispadias, kongenitální kloaky), abnormální poloha vzhledem k šourku penisu. Možné příčiny: faktory životního prostředí, plodová zúžení, chromozomální abnormality, genetické defekty v děloze.
 Příčiny anomálií plodu. Riziko malformací plodu
Příčiny anomálií plodu. Riziko malformací plodu Abnormality SOx genů a TVH Holt-Oram syndromu. Fibroblastové růstové faktory
Abnormality SOx genů a TVH Holt-Oram syndromu. Fibroblastové růstové faktory Co určuje dědičnost? Primární buňky zárodečné
Co určuje dědičnost? Primární buňky zárodečné Záložka pohlavních žláz. Vývoj zárodečných buněk
Záložka pohlavních žláz. Vývoj zárodečných buněk Záložka zárodečné buňky. stanovení chromozomální sex
Záložka zárodečné buňky. stanovení chromozomální sex Vývoj vnitřních pohlavních orgánů plodu. Záložka pohlavních žláz plodu
Vývoj vnitřních pohlavních orgánů plodu. Záložka pohlavních žláz plodu Diagnostika a léčení Turnerovým syndromem. Čistá gonadální dysgeneze
Diagnostika a léčení Turnerovým syndromem. Čistá gonadální dysgeneze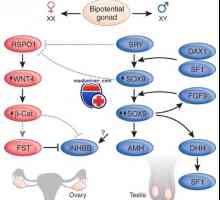 Mutace a genové duplikace dax1, sox9. pohlaví rozpor xy genotyp a kampomelicheskaya dysplazie
Mutace a genové duplikace dax1, sox9. pohlaví rozpor xy genotyp a kampomelicheskaya dysplazie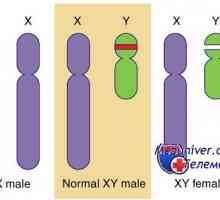 Genetické poruchy pohlavních žláz. Geny sry, WT1 a syndromy Fraser a Denis-dresha
Genetické poruchy pohlavních žláz. Geny sry, WT1 a syndromy Fraser a Denis-dresha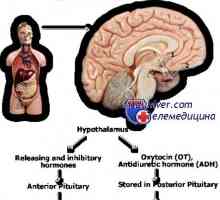 Mutace gonadotropin geny. Mutace v podjednotky LH a FSH
Mutace gonadotropin geny. Mutace v podjednotky LH a FSH Cysta-stop-genitální syndrom. Genetika mužské neplodnosti
Cysta-stop-genitální syndrom. Genetika mužské neplodnosti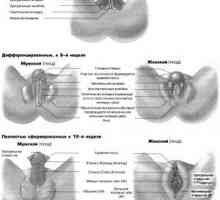 Vývoj pohlavních orgánů u plodu by týdny
Vývoj pohlavních orgánů u plodu by týdny Vrozené poruchy sexuální differentsirovkizabolevaniya způsobené chromozomálních abnormalit.…
Vrozené poruchy sexuální differentsirovkizabolevaniya způsobené chromozomálních abnormalit.…- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
 Pravda hermafroditismus
Pravda hermafroditismus Hypergonadotropní hypogonadismus
Hypergonadotropní hypogonadismus Primární hypogonadismus u mužů i žen: Léčba
Primární hypogonadismus u mužů i žen: Léčba Stanovení podlaha (spy jako určovací faktor varlete)
Stanovení podlaha (spy jako určovací faktor varlete) Karyotyp 46 xx žena
Karyotyp 46 xx žena Ovariální amenorea
Ovariální amenorea Vrozené vady pohlavních orgánů
Vrozené vady pohlavních orgánů