Galaktosemie
Video: Prevence genetické závady
galaktosemie - hereditární intolerance galaktózy. Toto onemocnění je způsobeno dědičné autozomálně recesivní porucha enzymem galaktóza-1-fosfát, který převádí uridiltransferazy galaktózu-1-fosfátu na glukózu-1-fosfát. Tato konverze je prováděna v buňkách jater, ledvin, mozku a v erytrocytech. Mechanismus zranění je spojen s akumulací galaktózy-1-fosfátu, které mají toxický účinek na tkáně.
Předpokládá se, že v oparu čočka závisí na vlivu galaktiola - látky vyrobené u pacientů s galaktózou a uloženého ve velkém množství v čočce a v jiných orgánech, je také vyloučeno močí. Klinicky rozlišit dvě formy - tvrdé, ostré a snadno, protragi-ment. Akutní forma se projevuje v prvních dnech po porodu.
Příjem příčiny mléko zvracení, průjem, zvětšená játra, žloutenka, doprovázené galakto-zuriey a aminokyselin, proteinurie. Do konce prvního měsíce existuje bilaterální katarakta. Dětí umírá v prvních měsících života ze selhání jater nebo interkurrentpyh infekčních chorob. Velmi důležité je včasná diagnóza nepřítomnosti galaktosy-1-fosfátu urndiltransferazy erytrocytů.
podle M. Lachmajer-Lutoslawska, A. Happe (1978), existují dvě možnosti galaktozemin: první charakterizována deficitem galaktóza-1-fosfátu uridiltransferazy druhý - nedostatek galaktokinázy. Druhé provedení odpovídá klinicky mírnou formou (bez vývoje zpoždění, bez jaterního onemocnění, ale s šedým zákalem). Prenatální diagnóza může být provedena na základě defektu enzymu v kultuře amniotických buněk. Pitva ukázala, cirhózu s ascitem, anasarca, žloutenka, krvácení do sliznic a vnitřních orgánů.
mozkové Změny a čočky nespecifické [Ivemark W. 1974]. Histologicky rozprašování obezita jaterní buňky až do vývoje cirhózy s uzlového tuku regenerátu, žlučovodu proliferace a proliferace pojivové tkáně.
Video: Proč děti špatně spí

Vrozené poruchy metabolismu aminokyselin
Vzhledem k úspěchu moderního biochemie V posledních dvou desetiletích byl schopen studovat patologii metabolismu téměř všech známých aminokyselin. Získané varianty těchto samostatných klinických jednotek metabolické onemocnění. Nicméně, patologické změny jsou popsány pouze v některých formách poruch metabolismu aminokyselin. Je třeba zdůraznit, že většina onemocnění metabolismu aminokyselin má charakteristickou morfologii, která je velmi obtížné pro ně postmortem diagnózu, ne-li předem disektor má klinické a biochemické data.
dědičně způsobené metabolickými poruchami nebo aminokyseliny spojené s vadou enzymů katalyzujících metabolické reakce určité intracelulární nebo vadné transportní proteiny nesoucí reabsorpci aminokyselin v renálních tubulech nebo procesů aminokyseliny absorpce kyseliny ve střevě. Onemocnění spojených s poruchou reabsorpcí v renálních tubulech byly volány tubulopatie, doprovázený porušením dopravy ve střevě - malabsorpce.
Při porušení meziproduktu metabolismu aminokyseliny tam giperaminatsidemiya více selektivní s giperaminatsiduriey- při porušování dopravních - často jen giperaminatsiduriya bez giperamiiatsidemii. Klinické projevy onemocnění metabolismu aminokyselin detekována převážně v kojeneckém věku a raném dětství.
Video: Tablety z kouření! Tabeks (Než začnete používat tento lék, přečtěte si návod)
V prvním roce života jsou většina náročné procesy CNS myelinizace, takže pokud by docházelo k porušování výměny jsou poškozeny na prvním místě. Giperaminatsidemiya vývojové giperampnatsidurii doprovázen onemocnění ledvin, ačkoli patogenní vztah mezi těmito jevy, není vždy dostatečně jasný. Porážka tubulární a intersticiální ledvin složka obvykle komplikována sekundární infekci s rozvojem pyelonefritidy. Zhoršená funkce ledvin dochází k narušení metabolismu fosforu (renální osteopatie) vápníku a, což je doprovázeno deformací kostí skeletu a poruchou procesu růstu.
To znamená, většina amino kyselin onemocnění metabolismu charakterizovaných poškozením mozku, ledvinách a kostry. Zpravidla se děti snadno dostat nemocných přidružené infekce.
 Glykogen spalničky skladování choroba, Andersen McArdl. Hersey nemoc, Thomson, kontejnery
Glykogen spalničky skladování choroba, Andersen McArdl. Hersey nemoc, Thomson, kontejnery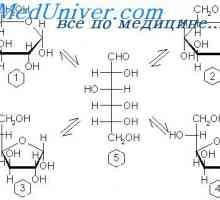 Fyziologie metabolismu glukózy. Transport glukózy přes buněčnou membránu
Fyziologie metabolismu glukózy. Transport glukózy přes buněčnou membránu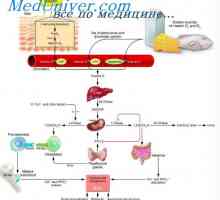 Fyziologie vitaminu D. Efekty a role vitaminu D
Fyziologie vitaminu D. Efekty a role vitaminu D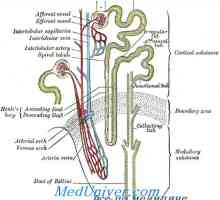 Izolace vápníku ledvinami. Izolace fosfáty ledvin
Izolace vápníku ledvinami. Izolace fosfáty ledvin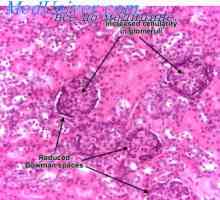 Léze v renálních tubulech. Glykosurie, acidaminuria, fosfátů
Léze v renálních tubulech. Glykosurie, acidaminuria, fosfátů Autosomálně recesivní polycystické onemocnění ledvin u dětí. Diagnostika a léčba
Autosomálně recesivní polycystické onemocnění ledvin u dětí. Diagnostika a léčba Novorozenecká žloutenka s metabolickými poruchami
Novorozenecká žloutenka s metabolickými poruchami Shvahmana-Diamond syndrom. Metafizariaya typ chondrodysplasia makové-Cusick a ukládání glykogenu
Shvahmana-Diamond syndrom. Metafizariaya typ chondrodysplasia makové-Cusick a ukládání glykogenu Renální glykosurie. Etiologie, patogeneze. Renální glykosurie vyvíjí v důsledku dědičné vady v…
Renální glykosurie. Etiologie, patogeneze. Renální glykosurie vyvíjí v důsledku dědičné vady v…- Fosfát-diabetem spojené dominantní X-vázaná onemocnění s hlubokým poškození výměnu vápníku a…
- Klindamycin (klindamycin). 7 hlordezoksiproizvodnoe linkomycinu. Synonyma: dalatsin q, klimitsin,…
- Poliestradiol fosfát (rolyestradiol fosfát). Polymerovaný ve vodě rozpustný lék estradiol fosfátu…
- Kodein fosfát (codeini phosphas). Synonyma: codeinum phosphoricum, kodein fosfát. Bílý krystalický…
- Urodan (urodanum). Směs má následující složení: 2,5 dílů piperazinu fosfátu, 8 dílů…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
 Porušení metabolismu aminokyselin
Porušení metabolismu aminokyselin Fanconiho syndrom: Příznaky, léčba
Fanconiho syndrom: Příznaky, léčba Metabolismus sacharidů u kojenců
Metabolismus sacharidů u kojenců Výživa a dědičnost dítě
Výživa a dědičnost dítě