Porušení lipidového metabolismu

Frekvence a klinický význam vede glykosfingolipidy narušení sdílení, které tvoří buněčné membrány mozku a většina ostatních tkání.
Video: Poruchy metabolismu lipidů a jeho korrektsiya- vše, co jste chtěli vědět [statiny / dyslipidemie].
Sphingolipid kompozice obsahuje sfingozinamino-alkohol, který má 18 zbytků uhlíku, na který jsou ostatní součásti připojené prostřednictvím aminové nebo hydroxylové skupiny. Deriváty sfingosin zahrnují ceramidy, cerebrosidy, sfingomyelin. Zejména rozdělení jednotlivých sfingolipidů, hladina enzymu bloku určení různých klinických jednotek. Takže sfingolipidozy s CNS lze rozdělit do dvou skupin:
- s primárním narušení metabolismu myelinu;
- s primárním porušení výměny cerebrosidy. Charakteristickým rysem hromadění hlavních typů sfingolipidy je třeba považovat za neurologickými příznaky, s výjimkou Fabryho choroba, některé formy Gaucherovy choroby a Niemann - Pick. K dispozici je devět subjektů, které odrážejí hromadění určitého sfingolipidu.
Společné pro všechny gangliosidy, jako podskupina sfingolipidy, musí být považovány za spojení ceramid přes zbytky boční hexózových N-acetylneuraminová (sialová) kyselin, a množství jeho hexózových skupin a skupin se liší. Gangliosidóza obvykle označován dopisy latiny, kde G - gangliozid- M, D, T - počet zbytků kyseliny sialové, v daném pořadí, 1, 2, 3. Čísla ve vzorci znamená počet molekul hexózy: 1 - tetra-, 2 - tri, 3 - di-hexóza. Všechny druhy gangliosidóza zděděná autozomálně recesivní.
GM1-gangliosidóza - nedostatek isoenzymů A, B a C, P-galaktosidázy s akumulací v tkáních M1 gangliosidu, asialoGM1 a gangiiozida-keratansulfát. Gen mapuje 3r21-r14.2. Když jsem typ onemocnění začíná při narození nebo krátce poté a končí s fatálními následky v prvních 2 let života. Hepatosplenomegalie a hluboký vývoj zpoždění psychomotorický v kombinaci se změnami kostí, hrubé rysy obličeje, které jsou typické pro MPC, hypertrofie dásní, makroglosie, vakuolizace krevních lymfocytů. Hrubě: rozšíření komor a mozkové atrofie. Mikroskopicky: akumulace a PAS-pozitivní materiál sudanofilnogo (gangliosidy) v neuronech a jejich následné smrti, glióza. V játrech, slezině, lymfatických uzlinách, brzlíku, kostní dřeně, ledvin - hojnost pěnových buněk.
U typu snížena isozymy II aktivitu a C P-galaktosidázu. Choroba začíná v 6-12 měsíců, osudovost vyskytuje ve věku 3-10 let. Klinický obraz je ovládán ataxii, psychomotorickou retardací, slepota. Somatické symptomy jsou prakticky nevyskytují. Morfologické změny podobné těm typu I, které se liší výskyt lézí mozečku neuronů, méně akumulace pěnových buněk ve vnitřních orgánech.
GM2-gangliosidóza Že obsahuje alespoň sedm formy. Takové potrubí je spojeno s mutacemi v různých lokusech genů, které regulují aktivitu hexosaminidázy A a B.
- Typ I (Thea nemoc - Sachs) - cm. „onemocnění centrálního nervového systému.“ Gen se mapuje na 15q23-Q24.
- Typ II (Sandhofa choroba) To způsobilo úplnou inhibici aktivity hexosaminidázy A a B. Tento gen mapován 5q13. Po proudu a v blízkosti klinických projevů Tay - Sachs, kromě toho, že je zvýšení jater, sleziny, ledvin. V nervové tkáně uloženy GM2 gangliosid a jeho asialovy analog, jak je v typu I. Mononukleární fagocyty z jater, sleziny, lmfouzlov, plic, nephrothelial hromadí neutrální glykolipid - globoside.
- Typ III CM2-gangliosidóza (Juvenile gangliosidóza) začíná ve 2-5 letech, smrt nastává před dosažením věku 15 let. Klinicky podobný typu I, biochemicky, částečné snížení ativnost hexosaminidáza A, akumulace gangliosidu omezen nervové tkáně.
- Typy IV-VII GM2 gangliosidóza To je extrémně vzácné, jsou popsány u dětí i dospělých.
GM3-gangliosidóza galaktozaminotransferazy enzym blokáda způsobil. Děti jsou nemocné při narození a umírají v prvních měsících života. velké množství gangliosidu hromadí v nervové tkáni a vnitřních orgánů.
Niemann-Pickova choroba (sfingomyelin-cholesterol lipidóza) Jedná se o autozomálně recesivní model dědičnosti. To zahrnuje skupinu onemocnění s hepatosplenomegalií a akumulaci sfingomyelinu, gangliosid a cholesterolu v mozku, na gangliové buňky, makrofágy, vnitřních orgánů. Z pěti klinických forem onemocnění (A, B, C, D, E) je nastavena na typy vad enzymů A a B, kde snížení sfingomyelinázy aktivitu. Gen mapuje 11r15.4- 15.1. Když styl s poruchou esterifikace cholesterolu. U typu III a IV není nainstalován primární Biochemická porucha. Jeden gen byl zmapován na 18q11, druhý - na 14q24.3.
- Typ A - akutní novorozenecké neuronopatická forma. Začíná v raném dětství do 6 měsíců objeví hepatosplenomegalie, zduření lymfatických uzlin, kůže má charakteristický hnědožlutý barvy, 50% pacientů ve fundu v oblasti makuly jasně třešňově červené skvrna (Příznak „třešeň jámy“), progresivní demence, ztratil motility , Smrt nastává v 3-4th letech života.
- Typ B - infantilní forma alelických variant zapojení typu A. CNS chybí.
- Typ C - neuronopatická juvenilní forma. Chronický průběh s poškozením mozku. Viscerální léze jsou podobné těm, které, když píšu.
- Typy D a E jsou klinicky podobné. Na rozdíl od příznaků prvního typu na 1-6 th let života, smrt nastává v období dospívání.
Ve všech formách vnitřních orgánů je uložena velké množství sfingomyelinu a jiných lipidů, které poskytuje charakteristickou žlutou barvu lymfatických uzlin a jater. Ve slezině na světlém pozadí jsou výrazně četné červeno-žluté folikuly. Označené cerebrální atrofie (s výjimkou pro typ B). Mikroskopicky: v orgánech s vysokým obsahem mononukleárních fagocytů velké pěnové buňky, cytoplazma, který je naplněn spoustou kapek, které neobarvené vzorky vypadá jako „moruše“. Tyto buňky se jednou, někdy i více jader. Hematoxylinem a eosinem barveny v cytoplazmě různých odstínů od šedé až hnědožluté, v závislosti na obsahu ceroid hnědého pigmentu (lipofuscin).
Mozek se uzavře, neurony oteklé s světle vakuoalizovanou cytoplasmě, kůra zdevastovala výrazné demyelinizaci bílé hmoty s gliózou a hromadění lipidů. Pěnové buňky se nacházejí v pia mater, mezery z Virchow - Robin, choroid plexus.
Sulfatidny lipidóza (jednobarevné leukodystrofie) - cm. „onemocnění centrálního nervového systému.“
Fabryho nemoc (trigeksoziltseramidny lipidóza) Má X-vázaná recesivní dědičnost. Vzhledem k deficitní lyzozomální -galakgozidazy gen, který mapuje Xq22.
Galaktosidázy A nedostatek vede k ukládání glykosfingolipidů především ve stěnách malých krevních cév téměř všech orgánů, stejně jako v rohovky epitelu, renální glomerulů a nephrothelial, kardiomyocytů, neurony, autonomní ganglia. akumulace lipidů ve vaskulárních výsledky endotelu ve vzhledu pěnových buněk, kde typické Zkosené lumen současně zvyšuje ektazie ovlivněn cévy. Pro diagnostiku in vivo je široce používán kožní biopsie. V polarizační mikroskopie odhalila usazenin v tkáních krystalické glykosfingolipidů mající podobu „maltézský kříž“.
To začíná v dětství a pomalu postupuje. Časné projevy patří opakující se záchvaty silné bolesti dolních končetin, parestézie, rozptýlených vaskulárních lézí na kůži, gipogidroz. Zvyšuje ledvin a selhání srdce, cévní mozkové příhody jsou možné.
Gaucherova choroba (glyukoziltseramidny lipidóza - typů I, II a III) Jedná se o autozomálně recesivní model dědičnosti. Mutantní gen mapovány na 1q21.
Metabolické poruchy a akumulace v těle glukosylceramid (glukocerebrosidu) v důsledku -D-glukocerebrosidasa aktivity snížení nebo absence v buňkách mesenchymálního původu.
Akumulace glukocerebrosidu v histiocytů jim dává charakteristický vzhled (Gaucherovy buňky). Tento velký průměr buňky od 20 do 100 mikrometrů s excentricky umístěné jádru a cytoplazmě nezhnofibrillyarnoy má tvar „zmačkaný hedvábný papír“ nebo „zmačkaný hedvábí“. Buňky slabě obarvené Sudan III, získá intenzivní PAS-reakce. Často pozorovány fagocytóza červených krvinek, bílých krvinek méně. Gaucherovy buňky se hromadí ve velkém množství, jako je slezina, lymfatické uzliny, brzlík, játra, kostní dřeň, stěny a adventitia cév vnitřních orgánů, prostorů Virchow - Robin. neurony v mozku neobsahují abnormální lipid, zároveň kontinuálně pozorována smrštění a smrti neuronů, gliózou a neuronophagia. „Goshepodobnye“ buňky se nacházejí u 30% pacientů s chronickou myeloidní leukémií, která je spojena se rozpadající fagocytózu leukemických buněk.
Video: Analýza testosteronu, kortisolu a obezity
Následný jsou tři klinické formy. Dospělý nebo chronické neuronopatického formu (typ I) je 80% všech případů a podobně Tay - Sachs nemoc a Niemann -pick distribuován převážně v Ashkenazi Židů. Toto onemocnění začíná v dětském věku se splenomegalií s hypersplenismem a pancytopenie, hepatomegalie, osteoporóza, patologické fraktury. V případě pozdního na začátku dlouhého období. Neurologické příznaky nejsou přítomny, ale histologicky v perivaskulárních prostorů Virchow - Robin nalezeno Gaucherovy buňky.
Infantilní nebo akutní neuronopatická forma (typ II) je 15% a je akutní, což vede ke smrti života 2 let. Dominují neurologické příznaky: extenzorového tuhosti, svalové hyper-, šilhání. V mozku - neuron smrti, reaktivní gliózu, perivaskulární akumulace Gaucherových buněk. Játra a slezina jsou zvětšené. Ta může trvat až 2/3 dutiny břišní.
Juvenilní nebo subakutní neuronopatická forma (typ III) - se nalézá především u jedinců ze Švédska. Objeví hepatosplenomegalie v 1. roce života, duševní poruchy se vyskytují ve věku 4-8 let. Průměrná délka života 15 let.
Farber nemoc (lipogranulematoz) Jedná se o autozomálně recesivní model dědičnosti. Gene není mapována. Nedostatek lysozomální kyselá tseramidazy doprovázeno hromaděním ceramidu a jiných gangliosidy kůže, lymfatických uzlin, centrálního nervového systému, vnitřních orgánů.
Klinické projevy: psychické a fyzické retardace, hrubé slabý hlas, srážky nastaví podkožní a periartikulární uzly, hepatosplenomegalie, zvětšení lymfatických uzlin, selhání srdce a plíce. Akumulace ceramidu a gangliosidy a tkání na rozdíl od jiných lipidóza vede nejen ke vzniku pěnových buněk, ale také pro rozvoj reaktivního granulomatózní procesu v přítomnosti granulomů lymfocytů, makrofágů, vícejaderných obrovských buněk a výsledku v fibrózou. Granuloma trvale lokalizovány v kůži kolem kloubů, krku, srdečních chlopní, pohrudnice. Pěnové buňky, PAS-pozitivní, obsahují komplexní lipidy.
Wolman nemoc (generalizované xanthomatosis) Jedná se o autozomálně recesivní model dědičnosti. Metabolická defekt - absence nebo snížení aktivity enzymu ve všech tkáních a buňkách jiných než erytrocytů s akumulací triglyceridů a esterů cholesterolu glykolipidy. Shromažďováním lipidy v lysozomech buněk dát pěnivá vzhled. ukládání lipidů je doprovázen zvětšení jater, sleziny, lymfatických uzlin. Charakteristika žluté barvy z jater, sleziny, lymfatických uzlin, nažloutlé pruhování střevní sliznice.
Patognomonické úbytek tuku v oblastech nekróz nadledvin velké množství vápenatých solí, které by mohly umožnit in vivo diagnózu radiograficky. Onemocnění začíná v prvních měsících života, smrt přichází do 6. měsíce života. Klinické projevy jsou velmi podobné těm, které v nemoci Niemann - Pick.
akumulace estery cholesterolu nemoc - když alelické formy aktivita kyseliny Wolman onemocnění lipáza částečně zachována. Významné změny - hepatomegalie jaterní fibróza, portální hypertenze, aterosklerózy. V kostní dřeni a střevní sliznici, může dojít k pěnové buňky.
 Pankreatitida vápník
Pankreatitida vápník Diagnóza sulfatidoza. Sfingomielinoz Niemann-Pickova choroba
Diagnóza sulfatidoza. Sfingomielinoz Niemann-Pickova choroba Galaktosemie
Galaktosemie Alzheimerova choroba: složek lipidů zabít nervové buňky
Alzheimerova choroba: složek lipidů zabít nervové buňky Metabolismus mozku. Regulace metabolismu mozku
Metabolismus mozku. Regulace metabolismu mozku Infarkt metabolismus v hypotyreózy. EKG v hypotyreózy
Infarkt metabolismus v hypotyreózy. EKG v hypotyreózy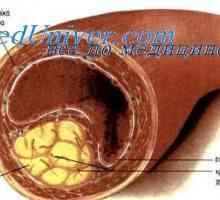 Hormonální příčiny aterosklerózy. Teorie a. Myasnikov koronární ateroskleróza
Hormonální příčiny aterosklerózy. Teorie a. Myasnikov koronární ateroskleróza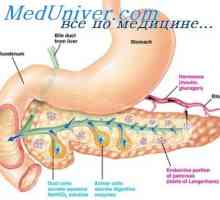 Diabetes tuku a libové. Svědění při cukrovce
Diabetes tuku a libové. Svědění při cukrovce Příčiny a mechanismy obezity. Hypotalamický obezita
Příčiny a mechanismy obezity. Hypotalamický obezita Charakteristika tuků (lipidů) a mastných kyselin
Charakteristika tuků (lipidů) a mastných kyselin Potřeby v tucích (lipidů) ve velmi předčasně narozených novorozenců
Potřeby v tucích (lipidů) ve velmi předčasně narozených novorozenců- Patogeneze peritonitida: porušením intraluminální mikrobiálního ekosystému, endogenní intoxikace
- Metodologické aspekty. Funkční uspořádání systému homeostázy klinických, antropometrických a…
- Gaucherova choroba se týká onemocnění, sfingolipidozam lipidov- akumulace v důsledku defektu genu…
- Hepatolentikulární degenerace (hepatolentikulární degenerace) -nasledstvennoe onemocnění vznikající…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Výborná lékařská encyklopedie IC nevronet. choroba
 Gaucherova nemoc, symptomy, léčba
Gaucherova nemoc, symptomy, léčba Niemann-Pickova nemoc
Niemann-Pickova nemoc Mucolipidosis
Mucolipidosis