Vrozené vady vývoje plic u dětí

Pozor: První pomoc při zlu vývoje plic, závisí na vás výrazy kliniky z subklinických symptomů kritickém stavu s cyanózou a tahidispnoe.
Cystická adenomatózní malformace
Zahrnuje řadu cystické a adenomatózní útvary ve frakcích jsou hamartomy s cystickou strukturou.
Vyloučení doprovázející poruchy orgánů (až 20%) s ledvinami, tenké střevo, brániční kýla, hydrocefalus, kosterní abnormality.
Prenatalnaya diagnóza polyhydramnios (komprese jícnu, nebo přímá zpráva cysty dýchacích cest) nebo hydrocephalus (srdeční selhání v důsledku zhoršené žilního návratu).
Diagnóza: RTG hrudníku - vidět více oddělené bubliny, často s hladinou kapaliny v rámci stejného podílu.
První pomoc: Chirurgická konzultace, plánování operace. Při určování prenatální diagnostika - co nejdříve.
pozor plicní hypertenze, zdravé plíce.
Vrozená lobární emfyzém
Vrozená lobární emfyzém: téměř vždy lékařské pomoci s pererazdutiem jednoho plicního laloku (bez emfyzématózní destrukci plicní tkáně).
Příčiny: malformace chrupavky, bronchiální strom, stenózy nebo vnější kompresní (cévní abnormality nebo nádor), polialveolyarnaya podíl.
Poznámka: Vždy vyloučit sliznice (meconium) konektor jako důvod (bronchoskopie).
Komplikace / problémy: posunutí, komprese a nadměrný průtok krve ve zdravém plicní tkáni.
První pomoc: Při monitorování provozu. vzácný případ nouzového provozu (lobektomie).
zabavení plic
Sekvestrace plic: cystická nebo homogenní oblast, zejména v nižších laloků, obvykle na levé straně. Tento nefunkční tkáň, která není ve spojení s bronchiálním systému. Nejčastěji se dodává s krví z aorty. Žilní odtok je implementován v systému nebo plicních žil. Vyvinutý z pomocného světla zárodek části předního střeva. Dříve vytvořenou vadu, miska snadné a zabavení jsou běžné pohrudnice.
Zvýšená pravděpodobnost spojených malformací.
Komplikace rozšířenou vlevo-vpravo zkrat, a tím srdeční insuficience (> 80% anastomózy - nádobami jícnu nebo žaludku fundu).
diagnostika Prenatalnaya: ultrazvuk.
Diagnóza, po narození rentgenového záření, je-li to nutné, CT angiografie (MR angiografie) a stskntigrafiya methylenchlori-difosfonát (akumulace v sekvestrace).
Léčba: metoda výběru - operace, i když ne kliniku kvůli riziku infekce. Před chirurgickým zákrokem - léčení klinického obrazu, resp.
Ageneze a aplazie plic
Bilaterální ageneze - extrémně vzácná vada neslučitelná se životem. Někdy chybí a průdušnice. Bronchiální tepny a žíly jsou obvykle chybí. To může být v kombinaci s jícnu vad, obličeje a asplenia. Proces, dochází k mnohem častěji v 50-60% pacientů má jiné vrozené vady :. srdce, urogenitálního systému, obratlů a žeber, brániční kýla, atd mírná kompenzační zvýšené. To je často zjištěno bronchiektázie.
Plicní hypoplazie (malé plíce)
Hypoplazie plic se vyskytuje u 10% dětských pitev v 85% případů spojených s dalšími vrozenými vadami. Klinicky charakterizuje vývoj SDR, závažnost, která závisí na stupni hypoplazii. Vrozená plicní hypoplazie může být primární nebo sekundární a idiopatická (statisticky významně vyšší), jedno a oboustranné. Primární plicní hypoplazie v důsledku genetických faktorů. To je popsáno v trisomie 13, 18 a 21, určité genetické syndromy dvojčat známých familiární případy.
Příčiny sekundární hypoplazie:
- abnormality cév zásobujících plíce (plicní chlopně Fallotova tetralogie et al.);
- oligohydramnion;
- nitrohrudní komprese plicní závaží (vrozená brániční kýla, vrozené hydrops plodu);
- komprese plic během hrudníku deformit (asphyxial hrudní dystrofie, těžká skolióza);
- Nedostatek dýchacích pohybů v děloze kvůli neuromuskulárních onemocnění.
Diagnóza idiopatické plicní hypoplazie dát v nepřítomnosti všech výše uvedených důvodů. Patologické anatomie obou forem hypoplazii stejného typu. Makroskopicky: obě plíce mohou být rovnoměrně snížen, nebo je výrazná asymetrie (např., Brániční kýla). Když hypoplazie je přímou příčinou smrti, nízká hmotnost je snížena o více než 40% a je často pouze 20-30% normální váhy pro gestační věk. Normálně donošených plodů a novorozenců nízká hmotnost -50 velký význam pro stanovení hypoplazii má index relativní hmot světla, tj, poměr hmotnosti plic na hmotnost plodu. Za normálních plodů a novorozenců 28 týdnech věku a starší, toto číslo je 0012, u předčasně narozených dětí až do 28 týdnů - 0,015 (tlumočení míru relativní množství světla, je třeba vzít v úvahu, že v přítomnosti patologických procesů v plicích, jako je zápal plic, BGM, aspirační syndromy a kol., relativní hmotnost je vyšší). V závislosti na mikroskopickém snímku jsou dvě hlavní formy plicní hypoplazie. První z nich - málo světla, ale na stupni zralosti těhotenství, i když se počet plicních sklípků je snížena, zatímco druhý - je výraznější nezralost plicní tkáně. Tato forma je v kombinaci s oligohydramniu. Je-li barva se speciálním detekovány v nepřítomnosti elastické tkáně interlobárních septa. Imunohistochemicky definovány podstatné snížení kolagenu typu IV. Morfometrické studie ukazují, že bez ohledu na příčinu v hypoplastické plicích sníží radiální alveolární počítat. Alveolární radiální skóre snadno určit na preparátů podle počtu alveolárních sept uspořádané na přímce tažené kolmo od terminálních bronchiolů do pohrudnice nebo lobulární septa. V normálním termínu kojence radiální alveolární skóre je 4,1-5,3. Další metodou stanovení hypoplazii - měření množství DNA jako indikátor plicních celkové buněčné populace plic.
hyperplazie plic
Často sekundární defekt. Zvýšení pozorované v plicní cystické adenomatózní vice plicní emfyzém vrozené jako vyrovnávací proces s jednostrannou agenezí nebo hypoplazii. Pravda hyperplazie - vždy obousměrný proces, způsobené obstrukcí horních cest dýchacích. Hyperplazie plic v kombinaci s hrtanu atrézie vyskytuje v autozomálně recesivní syndrom Fraser vyznačující Cryptophthalmos, abnormální uši, ledviny, Syndaktylie a kryptorchizmu. Makroskopicky: výrazná zvýšení plicní membrány se přemístí směrem dolů, světlo z otisku prstu hrany relativní zvýšení tělesné hmotnosti. Mikroskopicky: zralejší plicní tkáně se zvýšením alveolární povrchu ve srovnání s hmotnosti a gestační věk novorozence. Hyperplazie plic hrtanu atrézie je způsobena absencí výtoku kapaliny z plic do amnion dutiny v důsledku obstrukce.
Dodatečný světlo (tracheální další plic)
Tato velmi vzácná porucha způsobená porušení dělící primární bronchiální ledviny. Je nutné odlišovat od plicní sekvestraci a „průdušnice průdušky.“
podkovy plic
Je vzácná svěráky, v němž jsou plíce částečně spojené ve svých základnách a za srdce do přední části jícnu. Pokud žádné jiné vady jsou bez příznaků, ale to je popsáno ve spojení s různými cévními poruchami plic a jiných vad, jako je hypoplastickým pravé nebo levé plíci a Scimitaru syndromu.
Ektopická plicní tkáň
Možná ektopie plicní tkáně v krku, břicha a hrudní stěny, často spojená s kosterními abnormalitami a brániční kýly. Někdy ektopický plicní tkáně v dutině břišní se označuje jako ekstralobarnaya odlučování. Ektopie na krku je pozorován u iniontsefalii anomálií Klippel -Feyla a mňoukání. Příčiny mimoděložního plic není známa.
Heterotopie v plicích
Popsáno v podobě ostrovy heterotopie tkáni nadledvinek, slinivky břišní, štítné žlázy (bez C-buněk), jater a příčně pruhovaného svalstva, které se také nacházejí v plicní sekvestraci a hypoplastické plic. Jejich význam není velký, protože tyto heterotopie jsou pozorovány pouze u dětí s jiným těžkým zdravotním postižením, zejména srdce. Když anencephaly krevního řečiště po požití nebo se mohou dostat do plic ohniska gliální tkáně. V důsledku porodní trauma může nastat cévní embolie z tkáně mozečku, které jsou někdy mylný pro hamartomy nebo heterotopické tkáně.
Cystická plicní nemoc
Současná klasifikace plicní cystické onemocnění není ani zdaleka ideální. V současné době rozlišujeme vrozené a získané cysty. Získané cysty u kojenců může být důsledkem komprese bronchu infekce zvyšuje plicní tepny, přítomnost cizího tělesa v průduškách, bronchů hypoplazii. V této části nebudeme uvažovat o nich. K vrozené cysty patří bronchogenní cysty, plicní sekvestraci, cystická adenomatózní malformace plic, polialveolyarnaya podíl, kongenitální lobární emfyzém, cysty a limfangiomatoznye duplikace cystu.
Vrozené plicní cysty.
Vzácný malformace
Cysty jsou jedna nebo více, jsou vždy spojeny s bronchiálního stromu (porodu často naplněné tekutinou), jsou omezeny na jednu akcii.
Diferenciální diagnóza: pneumotorax.
Video: Newborn odstraněna část plic
Léčba: Chirurgie - metodou volby. Když napjaté cysty jako jejich nouzových opatření může být propíchnuta. Zvýšení velikosti a síly může dojít velmi rychle.
bronchogenní cysta - Toto vrozené cysty. v důsledku porušení primitivního střeva. Obvykle se nachází v mezihrudí blízkosti výběžku (51%), ale může být v pravém paratracheal oblasti podél jícnu, brány plicích nebo v různých jiných místech. Bronchogenní cysty jsou zřídka spojen s tracheobronchiálního stromu nebo umístěny v plicní tkáni. U kojenců jsou bez příznaků, nebo může být příčinou SDR. Hrubě: zaoblené cysta s hladkým nebo drsným vnitřním povrchem o průměru 1-4 cm, které je připojeno k tracheobronchiálního stromu, ale ne s ním spojeny. Obsah - clear serózní tekutiny v případů infekce - zakalený nebo krvavý. Mikroskopické cysty lemované řasinkami krychlový nebo cylindrického epitelu, někdy je skvamózní metaplazie. Stěna se skládá z malého množství pojivové tkáně, hladkého svalu, a chrupavky ložisek zřídka - bronchiálních žláz. Bronchogenní cysta v kombinaci s plicní sekvestraci, dalších plicních laloků popsaných v Downův syndrom.
Ekstralobarnaya zabavení diagnostikována u dětí mladších 1 měsíce. boys poměr: 4 dívky: 1. Silně: ostrov část světla s vlastním pohrudnice, často cystické změny. Umístěna kdekoliv - od krku na membránu, obvykle za levého dolního laloku, a defekt bránice často detekována. To může být umístěn v tloušťce membrány a osrdečníku. Asi 15% ekstralobarnyh izoluje lokalizované v peritoneální dutině, někdy spojen s jícnu nebo žaludku. Mikroskopicky: mezi normálními ložisek vytvořené alveolů jsou malé, ale moderní a obsahující chrupavky průdušky, v některých případech, bronchiektázie je výraznější, a alveoly jsou nedostatečně. Bronchioly a alveolární kanály nepravidelný tvar. Průdušky nejsou připojeny k tracheobronchiálního stromu. Lymfatické cévy jsou rozšířený, který se podobá vrozené lymphangiectasia. Tepen normální struktury, ale může se objevit i tenkostěnné nádoby. Vzhledem k absenci normálního bronchiálního odvodnění stagnuje v něm tajemství, které přispívá k infekci, tvorbu cyst a fibrózou. Ekstralobarnaya sekvestrace mohou být kombinovány s cystickou adenomatózní defektu plic, typy II a III. V nastat 65% případů malformace jiných orgánů, většina z nich vyznačuje brániční kýly, plicní hypoplazie, SPU a chonechondrosternon. TTP defekt - až do 6. týdne embryonálního vývoje.
na intralobarnoi zabavení abnormální část je obvykle umístěn v Posteromediální částech dolního laloku levé plíce mezi normální plicní tkáni a nejsou vymezeny z okolního parenchymu. Zřídka kombinovat s jinými PR. Velmi zřídka je diagnostikována u novorozenců. To se vyskytuje se stejnou frekvencí u chlapců a dívek. Makroskopicky: abnormální část vypadá atelektatichesky segment nebo cysty polycystické tkáně jsou naplněny bělavý nažloutlý průsvitný kapalině nebo rosolovité hmoty. Mikroskopické cysty jsou lemovány válcových nebo krychlový epitelu, plicní tkáň je nedostatečně. U starších dětí a dospělých v oblasti označené bronchiektazií chronického zánětu, fibrózy, cysty, na základě kterého mnozí autoři se domnívají, že ve starším věku, zpravidla v procesu nákupu, a vrozený zřídka.
Infantilní (vrozené) lobární emfyzém (vrozený velký podíl supertransparency) Je charakterizována protahování segment nebo lalok vzhledem k vnitřní nebo vnější bronchiální obstrukcí. Častější u chlapců. Klinicky diagnostikována u novorozenců a kojenců SDR, je extrémně vzácné - u starších dětí (nutno vyloučit získané rozedma plic), může být příčinou smrti. Hlavně ovlivnil horního laloku, zřídka - dva podíly. Způsobuje vady jsou: stenóza, atrézie, anomální vypouštění průdušek, poruchy ve vývoji bronchiální chrupavky, slizničních řas, mukózních zástrčky a odsát meconium v lumenu průdušek, obstrukce průdušek aberantních cév nebo bronchogenního cysty, atd. Tyto změny podporují hyperinflace oblast plic distálně od obstrukce splatnosti. tím, že je při vydechování méně vzduchu, než toky (ventilový mechanismus) odstraněny. Hrubě: ovlivněn podíl zvýšil, náhlý otok to prolapsy přes předním mediastinu směrem zdravé plíce, kompresi přilehlé oblasti. Mikroskopicky: viditelné rovnoměrně napjaté acini s plicních alveol a alveolů velikosti 3-10 krát více než standard, označený kontaktních přerušení v alveolárních sept. V některých případech je tento typ emfyzém způsobený zvýšením počtu alveolů (3-5 krát více než standard) v postižené laloku (polialveolyarnaya podíl), velikost alveolů se nezmění. Infantilní lobární emfyzém je často spojena s dalšími malformací, zejména srdce. Získal infantilní emfyzém pozorováno u předčasně narozených dětí, kteří byli na umělé plicní ventilace.
Vrozená vada cystická adenomatózní plic (fibrocystické dysplazie, adenomatózní plic, cystická hamartom adenomatózní plic) - Poměrně často (25% všech plicních CDF) gamartomatozny plic defektu, který se vyznačuje přítomností adenomatózních proliferujících cyst připomínající strukturu průdušek. Cysty jsou obvykle spojena s tracheobronchiálního stromu, cévní zásobení a žilní odvodnění realizován normálních plicních cév, s výjimkou kombinace s ekstralobarnymi zabavení. Malformace poprvé popsána Ch`in a Tang v roce 1949, Frekvence - 1. místo 25 000 až 35 Ltd. porod. Ve většině případů je diagnostikováno v prvních 6 měsících života, 70% - při narození nebo v 1. měsíci života, někdy i později. Téměř 90% všech v literatuře popsány pozorování se klinicky projevuje u dětí mladších 2 let. U novorozenců, 80% klinicky pozorovaných různým stupněm SDR, u starších dětí - přetrvávající nebo recidivující pneumonie. V současné době závada často diagnostikován prenatálně, s výhodou plodů 22,6 ± 3 týdny těhotenství, ale pozorování je popsáno v 5-8 týdenních plodů. Na prenatální diagnostiku této vady musí být odlišeny od brániční kýly, bronchogenního cyst, plicní sekvestraci a kongenitální lobární emfyzém.
Etiologie a patogeneze tohoto zlozvyku není znám. Předpokládá se, že narušení normální plicní zrání může být v důsledku atrézie nebo anomální bronchu jejich dělení. To vede k dysplazie plicní tkáně distální na lézi. Education dysplastická plicní tkáně plodu může být příčinou plicní hypoplazie nebo aplazie to. Na rozdíl od běžně rozvíjí plíce s cystickou adenomatózní dysplazie došlo ke zvýšení buněčné proliferace a snížení apoptózu. V nemocné tkáni, jak v fetální a neonatální zvýšené expresi několika růstových faktorů, zejména od destiček odvozeného růstového faktoru (PDGF-BB, TFR-BB), gliové odvozený neurotrofický faktor (GNTF -GDNF), o nichž je známo, že některé z faktorů stimulujících růst a vývoj plic, kde exprese GDNF v nemocné tkáni korelaci se stupněm proliferace. K dispozici je také rozpad zárodečného mezi parakrinní faktory.
V závislosti na klinických projevech, prognóze a morfologie rozlišit pět typů úhony.
- Typ 0 - acinárních dysplazie - malformace, který není slučitelný se životem. V kombinaci s srdečních vad a dermální dysplazie. Hrubě: plíce jsou malé, kompaktní, na řezy cysty podobají pokročilejší malé průdušky. Mikroskopicky: hojný mezi mesenchymu uspořádán malý (0,5 cm v průměru) bronhiolopodobnye konstrukce obloženy vysokou cylindrického epitelu se pohárkových buněk slizeprodutsiruyuschimi (slizniční diferenciaci pro tento typ unikátní vady) ve stěně jsou fibroznomyshechnoy chrupavky a žlázy.
- Typ I - cystická dysplazie - nejběžnější typ (50-75%). Diagnostikován u 1-tého týdne nebo měsíce života, ale může se objevit u starších dětí a dokonce i dospělé. Makroskopicky: jeden nebo více multilokulyarnyh velké cysty (průměr 10,3 cm) naplněné vzduchem a kapalinou, obklopené malým kartáčkem a zúženou normálního plicního parenchymu. Mikroskopicky: větší cysty lemován řasinkovým, psevdomnogo řádkem válcových a menší - kubické nebo cylindrického epitelu. V 1/3 případů v epitelu velkých cyst nebo bronhiolo-, alveolopodobnyh struktury v blízkosti největšího cysty jsou slizeprodu-tsiruyuschie buňky. Někdy cysty uvnitř a přiléhající struktury pozorovány bronhiolopodobnyh papilární proliferace epitelu, který se podobá bronchogenní karcinom. Cysty se skládá z elastického, kolagenu a tkáně hladkého svalstva, 5-10% případů zjištěn chrupavku. Typicky operovaných s příznivou prognózou.
- Typ II - střední - frekvence druhého typu (asi 20-25%). Pozorovat pouze v 1. roce života, ale má špatnou prognózu a častější kombinaci s jinými CDF -serdtsa, centrálního nervového systému, močového, pohybového aparátu, brániční kýla, některé z nich nejsou slučitelné s životem (např Arenas, Sirenomelia). Tento typ vady je pozorována při ekstralobarnoy sekvestraci. Makroskopicky: postiženou část nebo všechno světlo v příčném řezu na houbovitý a sestává z těsně vedle sebe s malým průměrem cysty od 0,5 do 2 cm, se připojují k průdušek a naplní vzduchem, když je dítě dýchání. Vnitřní povrch cyst hladké, lesklé nebo drsný. Jsou rovnoměrně rozděleny mezi plicní tkáně, sloučení s ním. Mikroskopicky cysty podobají pokročilé terminální bronchioly, respirační lemované kvádrových nebo cylindrického epitelu do zdi jsou vláknité, elastické, a hladkého svalstva vlákna. Mucinózní buňky a chrupavka chybí, ale v 5-10% případů může být ohniscích příčně pruhovaných svalů (rabdomiomatozny sub-option).
- Typ III - pevná - méně časté (5%) a téměř výhradně chlapci. Hydramnios těhotenství komplikována komprese rozšířené laloky jícnu nebo plic. Delší stlačení dolní duté žíly vznikne fetální hydrops. V krvi matky v 2. trimestru těhotenství, zvýšení počtu -feto proteinu. Makroskopicky: velké, husté, nádorové masy zabírají celý podíl nebo celou plic, vždy posunuté mediastina a plicních lézí často hypoplastický (cystickou hypoplazii plic). V části vidět malé dutiny připomínající cysty, zřídka více než 0,2 cm v průměru (s výjimkou pro rozptýlené, větší struktury bronhiolopodobnyh). Plicního parenchymu cysty je nerozvinutý. Mikroskopicky: postižené tkáně podobá nezralé, chybí průdušek plic. Nepravidelně tvarované hvězdicovité bronchiolo- jako struktury lemované kvádru epitelu a je obklopen alveolárních kanálků a plicních sklípků, také lemovány kvádrových epitelu. Slizničních buněk a chrupavka je nepřítomné. Cysty jsou pozorovány u výše popsaných typů chybí. Prognóza je závislá na procesu distribuce a množství uložené plicní tkáni, je stupeň dostupnosti a mediastinální pps posunutí. Úmrtnost je vysoká.
- Typ IV - periferní cystické distální acinárních - se vyskytuje u 2-10% případů. Chlapci a dívky jsou postiženy stejně často. Pozorována u kojenců a malých dětí v prvních 4 letech života. Makroskopicky: velké, multilokulyarnye, tenkostěnné, vzduch obsahující cysty se nacházejí na okraji plíce, a mohou komprimovat jiné orgány hrudníku, občas stává komplikované pneumotorax. Mikroskopicky, cysty lemované zploštělých alveolárního epitelu (alveolocytes typu I). Stěna sestává z husté mezenchymální tkáně, s jasně viditelné tepen a arteriol. Sliznice buňky, chrupavka a svaly chybí. Prognóza pro chirurgickou léčbou je příznivá.
Vrozená plicní lymphangiectasia (limfangiomatoznye cysty) K dispozici jsou primární a sekundární. Sekundární způsobeno obstrukcí lymfatickou či žilního odvodnění plic, primární - extrémně vzácné malformace. Ve 2 krát častěji postihuje chlapce. Důvody jsou heterogenní. Mohou být dědičné autozomálně recesivní, ale ve většině případů sporadická závada. Často spojena s jinými vrozené vady, zejména asplenií a onemocnění srdce, které byly popsány v Noonan syndrom, Turner a dolů. Primární plicní lymphangiectasia jsou izolovány (pouze postižené plíce) nebo projevem generalizované lymphangiectasia. U novorozenců tam klinicky odlišných SDR. Ve většině případů úmrtí nastává během prvních hodin nebo dnů života, ale občas mohou být první klinicky diagnostikována pouze u dospělých. Hrubě: plíce jsou zvětšené, tlustý, lumpy. Široké příčky a interlobárních pohrudnice viditelné za více cyst v průměru a 5 mm, s důrazem na frakční lehké konstrukce. V blízkosti brány do plicních cyst podlouhlými. Obsahové -limfa cysty, nebo v kombinaci s průdušky - vzduch a lymfy. Mikroskopické cysty se nachází v pojivové tkáni pod pohrudnice, v interlobárních septa, kolem bronchiolů a tepen. V sériových řezů ukazuje, že jsou součástí komplexní síť kanálů spojujících lymfu lišících se velikostí a postrádá ventilů. Cysty lemované zploštělého endotelu (endotelové markeru lymfatických cév jsou CD 31 antigeny a faktor 8). Tenká stěna jsou identifikovány elastické, kolagenová vlákna a zřídka - buněk hladkého svalstva. Absence obrovských buněk cizí tělesa ve stěně cysty rozlišuje patologie přetrvávající intersticiální emfyzém je obvykle komplikace mechanické ventilace u předčasně narozených dětí.
duplikace cysta - forma jednokomorovými duplikatsionnyh cyst gastrointestinální trakt. V hrudní dutině, které jsou umístěny v zadním mediastinu na pravé straně jsou připojeny k jícnu jsou zřídka spojen s průdušky. Mikroskopicky: cysta stěna lemována rozvrstvené z dlaždicových vzácně - žaludeční nebo enterickým epitelu. Žaludeční epitel může ulcerovat, což vede k cysty perforace. Duplikace cysty mohou být kombinovány s anomáliemi dolní krční a horní hrudní páteře.
Alveolární kapilární dysplazie - narušení struktury plicních cév. Vzácný smrtící patologie, který je příčinou vrozených plicní hypertenze, přetrvávající fetální oběhu a SDR u novorozenců. Mikroskopicky: proliferace pojivové tkáně a interlobárních interalveolar septa, ve kterém je snížen počet kapilár, je kontakt mezi alveolárního epitelu a kapilární offline. Plicní laloky jsou malé, radiální skóre snížit. Stěny malých tepen byly zesíleny v důsledku hypertrofie tkáně hladkého svalstva. Kromě toho, že je abnormální umístění plicních žil, které doprovázejí acinárních plicní tepny v srdci, ne v interlobárních septa, jak bylo pozorováno v normální plicní tkáni. Stěny žil abnormálně a umístěn zahuštěný. Důvody nejsou známy vadu. Sourozenci jsou zřídka ovlivněny. Dá se kombinovat s jinými poruchami zažívacího a močového traktu.
Video: Vrozené srdeční vady u dětí
Plicní arteriovenózní fistula - abnormální komunikace mezi tepny a žíly. Tento postup se ještě často lokalizovány v dolním laloku. Pozorována u 25% pacientů s onemocněním Osler - Weber - Rendu (rodina hemoragická teleangiektázie) - autozomálně dominantní dědičné onemocnění s vysokou penetrací. Homozygotní formě, v některých případech smrtících v raném dětství.
Vrozený deficit povrchově aktivní látky (vrozené alveolární proteinóza)
To je autosomálně recesivní dědičným deficitem jednoho z proteinů povrchově aktivní látky - povrchově aktivní protein B (SP-B), v důsledku mutací v kodonu 121 genu SP-B a jeho RNA - mRNA SP-B. Klinicky vyznačující se tím, že rychle progresivní respirační selhání ihned po narození. Hrubě: světlo hustá, vzrostla více než 2 krát, airless. Mikroskopicky: alveoly jsou rozšířené, lemované kvádru epitelu a naplněný eozinofilní, granulí, PAS pozitivních hmot s odlupujících alveolocytes a množství makrofágů. Posledním krokem je pozorováno ztluštění alveolárních stěn v důsledku proliferace fibroblastů. Imunohistochemicky definován nepřítomností nebo snížení proteinu, povrchově aktivní látky v normálním množství proteinu A a C. nepříznivou prognózou. Ve všech případech popsaných v literatuře, došlo k úmrtí během 1. roku života.
Primární plicní hypertenze
Hypertenze plicní cirkulace u novorozenců může být spojen s přetrváváním fetálních plicních tepen, hypertrofie svalové vrstvy, proliferaci a intimální fibróza někdy fibrinoidní nekrózu a arteritidy, což vede k větví obstrukce prekapilární plicní tepny a vznik sekundárních glomových anastomóz. V rámci této možnosti, nemoc se vyskytuje na bleskový typu dětí umírá v prvních měsících života. Často tam je náhlá smrt. Hypertrofie svalové membrány plicních cév (zejména malé vnutriatsinarnyh tepen) je také pozorován v plodu asfyxie, plicní hypoplazie, předčasné uzavření ductus arteriosus, herniace membrány.
Benigní familiární pneumotorax
U novorozenců je extrémně vzácné, nejčastěji vidět v mužských dospívajících. Popsal jako rodina s autosomálně dominantní a autozomálně recesivní typy dědičnosti.
 První pomoc při brániční kýly u novorozenců
První pomoc při brániční kýly u novorozenců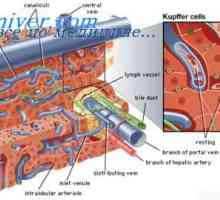 Patogeneze cystické adenomatózní vady plic. Vada světlo Typ I a II plod
Patogeneze cystické adenomatózní vady plic. Vada světlo Typ I a II plod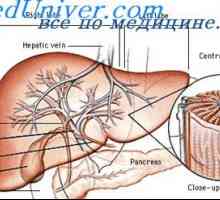 Cystická adenomatózní malformace plic. Příčiny plic vad u plodu
Cystická adenomatózní malformace plic. Příčiny plic vad u plodu Mechanismy hrtanu atrézii. Uzi hrtanu atrézie
Mechanismy hrtanu atrézii. Uzi hrtanu atrézie Vrozená vada bronchiolů v zárodku. Ultrazvuková diagnostika plicní vady u plodu
Vrozená vada bronchiolů v zárodku. Ultrazvuková diagnostika plicní vady u plodu Uzi s plicní sekvestrace u plodu. Prognóza a léčba plicní sekvestraci
Uzi s plicní sekvestrace u plodu. Prognóza a léčba plicní sekvestraci Fraser syndrom. Diagnostika a léčení syndromu u plodu Fraser
Fraser syndrom. Diagnostika a léčení syndromu u plodu Fraser Holt Oram syndrom. Gidroletalny syndrom
Holt Oram syndrom. Gidroletalny syndrom Frinsa syndrom. Diagnostiku a léčbu syndromu u plodu frinsa
Frinsa syndrom. Diagnostiku a léčbu syndromu u plodu frinsa- Resuscitační plicní edém
 Plicní edém. Mechanismy plicní edém
Plicní edém. Mechanismy plicní edém Pneumotorax
Pneumotorax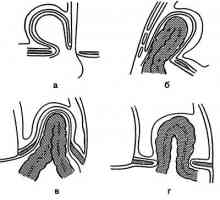 Diagnostika brániční kýla
Diagnostika brániční kýla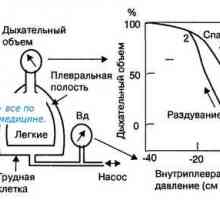 Faktory ovlivňující plicní inspirační objem ve fázi. Protažení plíce (plicní tkáně). Hystereze.
Faktory ovlivňující plicní inspirační objem ve fázi. Protažení plíce (plicní tkáně). Hystereze.- Pneumotorax u dětí v prvních dnech života dochází nejčastěji v důsledku protržení plicní tkáně…
- Vrozená brániční kýla. Tak vady ještě v děloze dochází břišní pohyb do hrudníku přes defekt v…
 Urgentní předlékařskou první pomoc při dušnosti doma
Urgentní předlékařskou první pomoc při dušnosti doma Emfyzém u novorozenců
Emfyzém u novorozenců Obstrukční léze gastrointestinálního traktu zažívacího traktu u dětí
Obstrukční léze gastrointestinálního traktu zažívacího traktu u dětí Syndrom mokré plic u novorozenců
Syndrom mokré plic u novorozenců Brániční kýla vada u novorozenců
Brániční kýla vada u novorozenců