Eponymní syndromy: Léčba

V tomto článku se podíváme na hlavních rysů titulních syndromy a jejich léčby.
Aaza syndrom. Klinická triáda vrozené anémie, trojitý šik palcem a měničem. Etiologie je neznámá.
Alfidi syndrom. Hypertenze v důsledku okluze celiakie kufru, což vede k obejít vedlejší průtok krve z pravé renální arterie. Zpočátku je popsáno jako ledvin viscerální ukrást syndrom.
Andersen syndrom. Triáda sestávající z periodické paralýzy, ventrikulární tachyarytmie a znaky dysmorfie (hypertelorismem, mikrognacii, nízko nasazené uši, rozštěp patra, malý vzrůst, skoliózy, Syndaktylie a clinodactyly). Paroxysmální paralýza může vyskytnout při hyper-, hypo- nebo normokalemia. Spojené s mutacemi v genu, který kóduje KCNJ2 k rychlým draslíkové kanály.
Barlow syndrom. Familiární forma PMC, které někdy je autosomálně dominantní rys. Geneticky heterogenní syndrom, vyznačující se prolapsu jednoho nebo obou z chlopně do levé síně během systoly. Poslech poslouchat srednesistolichesky tlačítkem myši a pozdní nebo pansystolic hluk. 20% jsou bez příznaků. Ženy trpí 2 krát častěji.
Barth syndrom. Mutace genu TAZ nachází na X chromozomu. To vede k dilatační kardiomyopatie, myopatie, nízký růst a neutropenie. Vylučování 3 metilglutakonikovoy vylučování kyseliny je téměř vždy dodrženy.
Beemer syndrom letální malformace. Vrozená fatální zdvojnásobení výstup pravé komory s hydrocefalem, těsnění kostí, trombocytopenie a abnormálnímu vývoji nosu.
Buyo syndrom. Jméno revmatické horečky podle autora. Buyo byl první, kdo zdůraznil, že je důležité zapojení do patologického procesu srdce v akutní fázi kloubní revmatické horečky.
Burnevilya onemocnění Pringle. Hamartom srdce a ledvin, v kombinaci s epilepsií, mentální retardace, kortikální hamartomy (tuberózní skleróza) a mazové adenomu. Může dojít k renální cysty nebo nádory.
Bradburi-Eggleston syndrom. Idiopatická autonomní poruchy, vyznačující se tím, posturální hypotenze, s častými projevy poruch funkce střev, močového měchýře, termoregulační a sexuální funkce.
Syndromem náhlého úmrtí. Jednou z hlavních příčin náhlého úmrtí mezi mladými lidmi v nepřítomnosti organických srdečních onemocnění, sekundárních proti SCN5A genové mutace. Narušena sodíkového kanálu, což vede ke vzniku a uchování komorových arytmií. Klinickým projevem BPNPG, elevace ST segmentu v vede V1-V3 a náhlou smrt nebo synkopa. Klinický fenotyp může být detekována události aymalina nebo prokainamid. Jedinou účinnou léčbou - implantace kardioverter-defibrilátor.
Carney syndrom. Také známý jako Carney komplexu. Tato kombinace myxom s fibrilací myxomy na jiných místech, například prsu, nebo kůže, a skvrnité pigmentace hyperaktivity žláz s vnitřní sekrecí, jako hypofýzy nebo varlat. Způsobena mutací v genu PRKAR1A chromozomu 17. syndrom se projevuje ve věku asi 30 let, ve většině případů, bilaterální myxomu, které, na rozdíl od ojedinělých případech se opakují po odstranění.
DiGeorgova syndrom. Poruchy vyplývající z vypuštění TVH1 genu na chromosomu 22q11.2, což vede k příštítné hypoplazii (a hypokalcemii), brzlíku hypoplazie (a nízké hladiny T-lymfocyty), abnormality v krvi výtokové části srdce, včetně tetrad Phaplu, cévní malformace páteř , přerušení oblouku aorty, aortální dekstrapozitsiyu, přidal podklíčkové tepny. U pacientů s tímto syndromem jsou obvykle přítomné mikrognatie, nízko nasazené uši, krátký nasolabiálních trojúhelník a malá ústa.
Shprinttsena syndrom také způsobila rozrušení ve stejném genu.
Dressler syndrom. Perikarditida spojená s infarktem myokardu, obvykle dochází po týdnu po začátku infarktu myokardu, ale může se také objevit několik měsíců později. Navrhnout autoimunitní původ v důsledku zpoždění rozvoje syndromu, přítomnost protilátek proti kardiomyocytů, přítomnost upravených lymfocytů a aktivace komplementu, časté recidivy spojené zánět pohrudnice, pleurální výpotek, stejně jako citlivost na NSAID a glukokortikoidy mohou být třecí šum perikard, horečka, perikardiální výpotek a pleurální abnormality intervagyuv PR, ST, T změna vlna, která je připomínající perikarditidy.
DMD. Zřetězeny s X-vázanou poruchy, na které neexistuje žádné zvláštní protein. Tam je výrazná slabost kosterního svalstva, které může maskovat dilatační kardiomyopatie. Tam je tendence k fibróze a zadnebazalnoy posterolaterální stěny levé komory. Supraventrikulární arytmie jsou častější než komorové fibrilace a AV bloku, které se vyskytují jako srdeční fibróza postupuje.
Ebsteinova anomálie. Malformace, přičemž pozorované patologické připevňovací chlopně trikuspidální chlopně vedoucí k posunutí ventilu směrem dolů. Z tohoto důvodu část pravé komory se nachází mezi atrioventrikulárního kruhu a umístit nástavec ventilu tak, že proximální část pravé komory je ve skutečnosti atrium a pravé komory dutiny se snižuje. Sledování dysplazie trikuspidální chlopně. Závažnosti stavu, spojeného s stenózou nebo plicní neprůchodností, a s ASD a měniče.
Eisenmenger syndrom. Defekt s plicní hypertenzí a cyanóza. To nastane, když se tlak v pravé polovině srdce se stává větší než vlevo, a tam je žilní vypouštění krve do tepenného řečiště. Poprvé byl popsán v roce 1897 z 32-letého muže s měničem.
Syndrom Ellis van Creveld (Hondroektodermalnaya dysplazie). Autozomálně recesivní onemocnění charakterizované malého vzrůstu způsobené metafýzy dysplazie, polydaktylií, dysplazie nehty a zuby, a obvykle ASD. Koarktace aorty, hypoplastických levé srdeční komory a zpět klesající tepna se vyskytuje u 20% případů.
Svalová dystrofie, Emery-Dreifuss. Klinicky se projevuje triády sestávající z prvních kontrakcí lokte, Achillovy šlachy a zadní krční svaly, progresivní myopatie kosterního svalstva, a srdeční projevy. Za prvé, zahrnují sinusovou bradykardii, AF a flutter síní, atrioventrikulární blok pak vznikají velmi konstantní VT a ventrikulární fibrilaci. Může také dojít k srdeční zástavu. Často náhlá smrt před dosažením věku 50 let. Tato nemoc je spojena s X chromozóm. Je to spojeno s genem, který je zodpovědný za kódující jadernou membránový protein zvaný emerin.
Fallotova tetralogie. Kombinace stenózy plicní tepny, VSD, dekstrapozitsii aorty a hypertrofie pravé komory, která způsobuje cyanózu v novorozeneckém Fallotova tetralogie je 10% všech vrozených vad a je častější u lidí, včetně plicní chlopně defektu síňového septa a neporušený interventrikulárního peregorodku- pentád Fallotova - tetralogy a ASD nebo PFO.
Friedreichova ataxie. Degenerativní onemocnění mozku a míchy, vyznačující se tím spinální ataxie, kosterní deformace, dysartrie, a kardiomyopatie. Často je hypertrofie levé komory, stejně jako asymetrické hypertrofie oddílu. Nejméně - dilatační kardiomyopatie. Může být fibrilace síní. To je autosomálně dominantní znak, s mutací označené jako nestabilní GAA trinukleotidové (guanin-adenin-adenin), zjištěného v prvním intronu genu frataxin na chromozomu 9q13.
Friedreichova ataxie nemoc. Náhlé zhroucení krčních žil, které byly dříve rozšířených na každém diastoly, protože lepicí perikardu. Také známý jako mediastinoperikardit nebo podepsat Fridreyha.Sindrom Holt-Oram. Autosomální dominantní poruchy, také známý jako srdce straně syndrom, který se střetává s růstem horních končetin, obvykle spojené se sekundárním ASD, ale také s měničem, PMK, ductus arteriosus. Deformace horní končetiny, může být sotva znatelné (např., Distálně umístěna trehfalangovy nebo palec), nebo výrazné (klíční kost a hypoplazie phocomelia).
Heidi syndrom. Kombinace gastrointestinálního krvácení a aortální chlopně s kaptsifikatsiey. V souladu s původním popisem ve 1958 Heidi, při krvácení došlo vlivem získaných von Willebrandův choroby typu 2a, způsobené vysokým tlakem v mezikruží aortální chlopně, což vede ke krvácení arteriovenózních zkratů ve střevě. Krvácení se zastaví, když se protéza ventil.
Hurler syndrom. Autozomálně recesivní porucha vede k akumulaci deficitu mukopolysacharidy lysozomálního enzymu alfa-biduronata. To je také definována jako mukopolysacharidóza IH type (MPSlhi). Klinické projevy zahrnují hrubé obličejové rysy, zákalu rohovky, gepatosplenomegapiyu, ztluštění kůže, mentální retardace a srdeční patologie. To zahrnuje restriktivní kardiomyopatie v důsledku endomyokardiální fibroelastóza, zúžení koronárních tepen, zahušťování chlopně (vlevo více než vpravo) a selhání. Nejvíce umírají během prvních deseti let. Hunter syndrom (MPS II) má pomalejší průtok. Krku syndrom (MPS) je nejvhodnější pro mukopolysacharidózy.
Syndrom Jervell-Lange-Nielsen. Autozomálně recesivní zabolevaenie doprovázen hluchotou, způsobené mutací v genu KVLQT1 nebo KCNE1 gen zodpovědný za pomalých draslíkových kanálů se podílejí na tvorbě akčního potenciálu. Jako výsledek intervalu QT je prodloužen, je zde riziko, typu ventrikulární tachykardie „piruety“ a náhlé srdeční smrti.
Kartagener syndrom. Klinický triáda situs inversus, patologie čelních dutin a ještě řasinek z řasinkami epitelu. Pacienti pozorována opakující se infekce dýchacích cest, zánět vedlejších nosních dutin, bronchiektázie a neplodnosti mohou být některé ztráta čichu nebo nízké hladiny IgA. Dědičnost je autozomálně recesivní. Defekt je v genu, který kóduje protein dynein, který určuje strukturu ciliárního epitelu. Také známý jako SIEVERT syndrom.
Kawasakiho nemoc. Akutní vaskulitidou, která postihuje děti a projevuje se horečkou, krční lymfadenopatie bilaterální zánět spojivek, zarudnutí nebo změnu měřítka padoney a nohou, aneurysma koronární tepny nebo ektázie. To může vést k infarktu myokardu a náhlé smrti. Etiologie je neznámá.
Kearns-Sayre syndrom. Klinické triáda se skládá z AV bloku, pigmentového retinopatie a chronické progresivní externí oftalmoplegie. Toto onemocnění je způsobeno delecí několika mitochondriálních genů. Ve většině případů, je dannoezabolevanie izolovaným jevem a není dědičná.
zrakového nervu dědičná neuropatie Leberova. Mitochondriální encephalomyopathies charakterizované bezbolestné ztrátou zraku v mladém věku. To může být doprovázeno krátkým intervalem PR a předčasné vzrušení.
Lenegre choroby Lev. To je také známé jako progresivní familiární typu I AV bloku. Autosomální dominantní poruchy spojené s chromozomu 19, které jsou příznaky raménka blokového blokády, QRS komplexy jsou široké, který může postupovat k úplné blokády. Nemoc je charakterizována progresivní familiární blokádou typu II, tak, že tyto jsou úzké ORS komplexy. K dispozici je zrychlený degenerativní proces, který má vliv především na vodivosti.
Loefflerův syndrom. Vzácná forma endokarditida, následovaný vysoké hladiny cirkulujících eosinofilů. Hlavní příčinou může být infekce způsobená hlístů, nebo s leukémií, ale ve většině případů je hlavní příčina není známa. Zpravidla rentgenového světlo rozptýlené uzlu zatemnění. Akutní forma charakterizována eozinofilní vaskulitida, což vede k expanzi srdečních dutin, vzhledem k tomu, chronické formy způsobuje fibrózu myokardu, což vede ke klinické syndromu restriktivní kardiomyopatie, díky které snížené toleranci zátěže, tam stridor, hepatomegalie, atrioventrikulární blok, selhání mitrální a trikuspidální trombóza ventil.
Syndrom Lown-Ganong-Levine. Fenomén předčasné komorové charakterizované krátkém intervalu PR (méně než 120 ms) a dobu trvání komplexu QRS normální, následované záchvaty SVT, ale bez AF nebo flutter síní. U pacientů bez anamnézy zvýšeného tachykardie atrioventrikulární vedení. Lown, Ganong a Levin soobshili o tento syndrom v roce 1952, i když se poprvé byl tento syndrom popsán jako úředník v roce 1938 nebyl zjištěn strukturní abnormality, které by byly příčinou syndromu Lown-Ganong-Levine. Nemoc může dojít v důsledku Intrasite parauzlovyh vlákna nebo obcházet atrioventrikulární uzel. Nicméně, většina pacientů jiné příčiny paroxysmální tachykardie byla detekována elektrofyziologické vyšetření, jako je například AVURT a ne obejít atrioventrikulární uzel. Proto syndrom Lown-Ganong-Levine - syndrom související s doelektrofiziologicheskoy epochy studie popisující paroxysmální tachykardie krátký PR intervalu, který může být na spodní hranici normálu (2-4% dospělých PR intervalu méně než 120 ms).
Libman-Pytle syndrom. Projevem systémový lupus erythematodes srdce, která se vyskytuje v pozdní fázi onemocnění, a nachází se v 50% pacientů po smrti. Vyznačující se sterilní Warty léze klapky a akordy, včetně fibrinu, neutrofilů, lymfocytů a histiocyty. Obvykle Užaslý mitrální a aortální chlopně, i když ve většině případů nejsou žádné klinické příznaky. Je častější chlopenní nedostatečnosti než chlopně. Takové poškození může dojít s antifosfolipidovým syndromem. Toto onemocnění náchylnější ženy.
Lutembacher syndrom. Kombinace mitrální stenózy (vrozenou nebo získanou) a defektu síňového septa (vrozený nebo iatrogenní), s bočníku zleva doprava. V případě, že ASD je velký, plicní hypertenze může být zabráněno v důsledku dilatace pravého srdce.
Marfanův syndrom. Poruchy vyplývající z mutací v genu FBN1 na chromosomu 15q21.1, fibrillin-1 kódování tvořící mikrovláken, které tvoří extracelulární matrici. Některé z charakteristických rysů syndromu - s vysokým růstem, kyfóza, skoliózy, trychtýř hruď deformita, vzhůru posunutí oční čočky, rozšíření mozkových plen, odchlípení sítnice, a více srdeční abnormality (prolaps mitrální a insuficience, dilatačních dutin, dilatace kořene aorty se selháním a zvýšené riziko rozvrstvení, arytmie). Pacienti mohou být zvýšené riziko endokarditidy sekundární k valvulární patologie. 75% se dědí autosomálně dominantní, ostatní - jsou izolované případy.
Morquio syndrom. Jeden z poruchy doprovázené hromaděním mukopolysacharidů - mukopolysacharidóza zadají IV B, dědičné autozomálně recesivní způsobem. Existují dva typy. Typ A je způsobena deficitem galaktosamin-6-sulfatázy, zatímco typ B je způsobena deficitem beta-galaktosidázy. Klinické projevy zahrnují malý vzrůst, kosterní změny a narušení kloubů, zákalu rohovky, gepatomegapiyu, aortální a mitrální insuficience. Srdeční selhání může být způsobeno infiltrative kardiomyopatií nebo v důsledku selhání.
Noonan syndrom. Dysmorphic syndrom charakterizovaný srdečními abnormalitami, malého vzrůstu, nízko nasazené uši, hypertelorismus, hluchota a hemoragická diatéza. Toto onemocnění se dědí autosomálně dominantním způsobem. Srdeční problémy zahrnují plicní chlopně. Tento syndrom je někdy nazýván „mužský Turnerův syndrom,“ ačkoli to postihuje obě pohlaví a nemá žádné chromozomální aberace.
Ortner syndrom. Zpočátku bylo popsáno jako opakující se stlačení nervů pokročilý hrtanu levé síně, když E trální zúžení způsobuje chrapot kvůli hlasivek obrnou. To je někdy používán k popisu jakéhokoliv benigní srdce nebo nitrohrudní proces, který má vliv na návrat laryngeální nerv. Levý nerv je postižena častěji než napravo vzhledem k zvláštní situaci ve vztahu k oblouku aorty.
Angina (varianta) Prinzmetalova. Vyskytuje se jako výsledek záchvatu věnčitých tepen a může vést k AV bloku a IM. Mohou být zabráněno tím, že blokátory vápníkového kanálu zpomalit s dlouhým účinkem.
Římsko-Ward syndrom. Autozomálně dominantní stav způsobený mutací v genech chromozomů 3, 4, 7, 11 a 21, které kódují různé složky jsou sodné a draselné kanály. Interval QT je prodloužen, existuje vysoké riziko vzniku typu tachykardie torsades de pointes a náhlou smrt. Ten není doprovázeno hluchotou, a tím se liší od syndromu Jervell-Lange-Nielsen.
Shprinttsena syndrom. Nemoc je způsobena mutací v genu TVH1, který je také zodpovědný za syndrom DiGeorgeova. Speciální funkce: - srdeční anomálie (obvykle VSD), rozštěp patra, mentální retardace a typická tvář s výrazným nosem, úzké oční štěrbiny a mikrognacií. Jak je dobře známo, jak patrová kardiofatsialny syndrom.
Stokes-Adams syndrom. Synkopa indukované arytmie. Také známý jako syndrom, Morgagni Spence syndromu.
Sydenhamova chorea. Pozdní projevy revmatické horečky v důsledku zánětlivé reakce způsobené autoprotilátkami v bazálních gangliích, nucleus caudatus a po skupině A streptokokové infekce obvykle začíná 3 měsíců po primární infekci, symptomy mohou přetrvávat po dobu 2 týdnů. Je charakterizována tím, mimovolné pohyby svalů nekoordinace pohybu a emoční nestabilitu.
Taussig-Bing syndrom. Vrozená vada.
Tietze syndrom. Žebro chrupavka zánět nejasné etiologie. Vyznačující se tím, nádor chrupavky, která odděluje syndrom z jiných typů zánětu chrupavkami. Nádor může zůstat po úlevu od bolesti. s výhradou k mužům v jejich počátku třicátých let hlavně nemoci.
Townes-Brox syndrom. Autozomálně dominantní stav, kombinující anální atrézie, renální patologie, horní, dolní končetiny a uši, a v některých případech doprovázeny srdečních vad včetně defektu síňového septa a měniče.
Turner syndrom. Onemocnění, která vyplývá z nepřítomnosti jednoho z chromosomu X, CW. Jeho vlastnosti jsou koarktace aorty, malý vzrůst, absence sekundárních pohlavních znaků, krční pterygia, valgus deformity loketního kloubu a lymfatického edému. Toto onemocnění je jednou z nejčastějších chromozomálních mutací.
Tvidlera syndrom. Není striktní eponym. Fenomén permanentního kardiostimulátoru poruchy způsobené vystavením pacienta ke generátoru pulsů.
Wenckebach srdce. Popis srdce, který je umístěn na středové čáře, a má menší velikost. Stejná nemoc známá jako mesocardium nebo „visící srdce.“
Wenckebach Fenomén. Forma II Stupeň atrioventrikulární blok, vyznačující se tím, progresivní prodloužení intervalu EKG Pra až do fibrilace buzení není drženo v komorách.
Williams syndrom. Nadkpapanny vrozená aortální stenóza se stenózou obvodovou plicnici, hyperkalcemie, elf tvář, neschopnost vlastní identifikace, mentální retardace, strabismu a abnormality zubů. Levá komora může být zbytnělé a dutiny mohou být rozšířené. Dále, koronární arterie může být také rozšířen nebo zvlněný, a urychlit aterosklerotické proces. Tito pacienti mají vyšší riziko endokarditidy a náhlé smrti. Tam je autozomálně dominantní přenos, v případě, že zdědil syndrom.
Wolff-Parkinson-White syndrom. Arytmie, které vznikají v důsledku dodatečného atrioventrikulárního způsobem. Vlastnosti EKG: sinusový rytmus, PR intervaly méně než 120 ms, komplex QRS je větší o více než 120 ms, vzhledem k D-vlny (pre-excitace), která vznikla v důsledku antegrádní nesoucí na další vodivé cestě. Pacienti mohou být nestabilní chování touto cestou, která vede ke změnám v EKG.
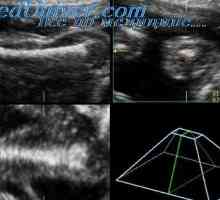 Kampomelicheskaya dysplazie plodu. Diagnóza kampomelicheskoy dysplazie
Kampomelicheskaya dysplazie plodu. Diagnóza kampomelicheskoy dysplazie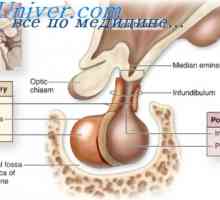 Vrozené zkrácení femuru. Štěpení rukou a nohou plodu
Vrozené zkrácení femuru. Štěpení rukou a nohou plodu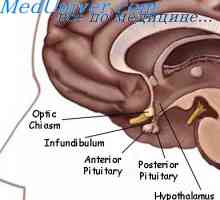 Dehet syndromu u plodu. Aase syndrom, Holt-Oram plod
Dehet syndromu u plodu. Aase syndrom, Holt-Oram plod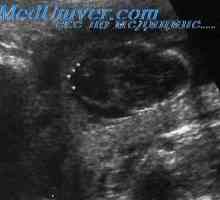 Vater Association u plodu. Polydaktylie syndrom a fetální goldenhara
Vater Association u plodu. Polydaktylie syndrom a fetální goldenhara Akromezomelicheskaya dysplazie. eykardi syndrom
Akromezomelicheskaya dysplazie. eykardi syndrom Holt Oram syndrom. Gidroletalny syndrom
Holt Oram syndrom. Gidroletalny syndrom Noonan syndrom u plodu. sekvenční oligohydramnion
Noonan syndrom u plodu. sekvenční oligohydramnion Pfeiffer syndrom. Diagnostika a prognóza Pfeiffer syndromu
Pfeiffer syndrom. Diagnostika a prognóza Pfeiffer syndromu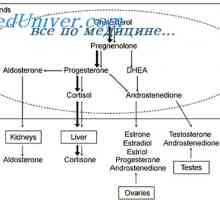 Trombocytopenie syndromy. Poloměr aplasia syndrom
Trombocytopenie syndromy. Poloměr aplasia syndrom Pierre Robin syndrom. Septo-optická dysplazie plodu
Pierre Robin syndrom. Septo-optická dysplazie plodu Trizomie 13 Patau syndrom. Přidružené anomálie Patauův syndrom
Trizomie 13 Patau syndrom. Přidružené anomálie Patauův syndrom Anomálie ženských pohlavních orgánů. Syndromy Kaufman-mák-Cusick a Mayer-Rokitansky-Kuster-Hauser
Anomálie ženských pohlavních orgánů. Syndromy Kaufman-mák-Cusick a Mayer-Rokitansky-Kuster-Hauser Cysta-stop-genitální syndrom. Genetika mužské neplodnosti
Cysta-stop-genitální syndrom. Genetika mužské neplodnosti Dědičné neutropenie. Těžká kongenitální neutropenie syndrom Kostmann
Dědičné neutropenie. Těžká kongenitální neutropenie syndrom Kostmann Occlusion větví oblouku aorty vede k mozkové ischémie a horních končetin. Etiologie aterosklerózy,…
Occlusion větví oblouku aorty vede k mozkové ischémie a horních končetin. Etiologie aterosklerózy,…- Okluze proximálních podklíčkové tepny podklíčkové tepny v důsledku nástěnné malby na místě původu…
- Smíšené onemocnění pojivové tkáně (Sharp syndrom), vyznačující se kombinací jednotlivých prvků…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
 Monogenní syndromy mvpr
Monogenní syndromy mvpr Mvpr syndromy. Chromozomální a genetické syndromy
Mvpr syndromy. Chromozomální a genetické syndromy