Syndromem mnohočetné endokrinní neoplazie, typu I (ME 1): příčiny, příznaky, léčba, symptomy
: příčiny, příznaky, léčba, příznaky")
MEN 1 (nebo Wermer syndrom) dědí autosomálně dominantním způsobem a nachází se s frekvencí 2-20 případů na 100,000 populace. Přibližně 10% z mutací v genech, které jsou základem tohoto syndromu, jsou de novo- takové případy se nazývají sporadické.
MEN 1 se liší v celé řadě klinických příznaků, které se projevují v různých rodinách s různou frekvencí.
Nejběžnější hyperparatyreózy, který v té či oné období života se vyskytuje v 95-100% nosičů genetické vady. Diagnostická kritéria pro hyperparatyroidismem v těchto případech se neliší od těch v sporadické onemocnění. Obvykle (ale ne vždy), že hyperparatyreóza je prvním klinickým projevem MEN 1. Na základě hyperparatyreózy je hyperplazie příštítných tělísek. Tyto žlázy nejsou vždy rostou najednou, ale odpuštění hyperparatyreózy může poskytnout pouze resekci téměř všech příštítných tělísek tkáně. MEN 1 je příčinou hyperparatyreózy pouze 2-4% z celkového počtu pacientů s tímto onemocněním.
Mezi pankreatických nádorů a gastrointestinálního traktu u mužů 1 gastrinom tvoří 20-60% [typicky způsobí Zollinger-Ellisonův syndrom (PPA)]. PPA se nalézá přibližně u 25% pacientů z rodin s muži 1. více než 20% nádorů buněk ostrůvků představují inzulínu, a ostatní - sdílet provozní (sekretující glukagon nebo TTI) nebo spící nádory. Gastrinom s MEN 1, jsou obecně malé velikosti, vyznačující se tím, multicentrické růstu a často lokalizované mimo slinivky břišní (typicky pod sliznice dvanácterníku). Gastrinom s MEN 1 (například v ojedinělých případech), jsou často maligní, ale z neznámých důvodů, které jsou méně agresivní než sporadických nádorů.
gastrinom Diagnóza je založena na detekci hypergastrinémie v kombinaci se zvýšenou sekrecí kyseliny chlorovodíkové v žaludku. Ten umožňuje vyloučit další častější příčiny hypergastrinémie (např achlorhydrií). V případě pochybností, lze použít vzorek se látka, která stimuluje sekreci gastrinu nádoru, ale ne normální tkáň. blokátory histaminových receptorů a inhibitory protonové pumpy reflexní zvýšení hladiny sérového gastrinu, a měl by zrušit respektive 30 hodin a 7 dní před diagnostické stanovení hormonu. Někteří autoři považován za spolehlivější diagnostickou techniku definici střevní hormony po standardizované snídani.
Vzhledem k malé velikosti gastrinu odhalit jejich lokalizace je obtížné. CT a MRI v takových případech nejsou informativní (i když to může odhalit metastázy v játrech). Lokalizace gastrinu nejčastěji detekována intraoperační a endoskopickou ultrazvukem selektivní injekci intraarteriální sekretin (a následným stanovením gastrinu v jaterních žil krvi) a skenování se značeným oktreotidu. Přesto téměř polovina případů stanovit lokalizaci gastrinom před selhání operace.
Inzulinom na MEN 1 je diagnostikována běžnými biochemickými studiemi. Multicentrický růst takových nádorů je také obtížné odhalit. Někdy to může být provedeno s použitím endoskopického ultrazvuku a selektivní infuzi intraarteriální vápníku k definici inzulínu v hepatické žíle krvi.
Adenomy hypofýzy se nacházejí v 25% nosičů genetické vady. Většina z těchto adenomů vylučujících prolaktin, růstový hormon (GH) nebo bez ní, a následně tím, že vylučuje nádorů pouze GR, nefunkční adenomů a ty, které vylučují velké množství ACTH (Cushingova nemoc).
Jedno z provedení MEN 1, vyznačující se tím, zvýšenou mírou prolaktinu, karcinoidní nádory a hyperparatyreózy a vzácnosti gastrinu, ale kde gen mutace je zřejmě liší od charakteristik jiných provedeních syndromu. tumorů hypofýzy obvykle představují makroadenomu. Jsou zřídka maligní, ale rostou mnohem agresivnější a méně citlivý na lékovou terapii než v ojedinělých případech. Diagnostika a léčba se nelišily od těch v sporadických adenomů.
Když MEN 1 nadledvin adenom někdy detekována. V takových případech je obtížné rozhodnout, zda je Cushingův je způsobeno primárního nádoru adrenální syndromu, basofilní adenomu hypofýzy nebo ektopických sekrece ACTH. Nejčastěji hyperkortizolizmus je však výsledkem nádoru hypofýzy. Nadledvin adenom často doprovázeny Islet nádory slinivky břišní. Je popsán jako 7 případy feochromocytom, ve kterých byly nalezeny mutace, které jsou typické pro MEN 1, MEN a ne 1I. Na MEN 1 pozorováno onemocnění štítné žlázy, ale jejich spojení s tímto syndromem podložena. V 30-90% pacientů s MEN 1 rodiny pozorovány podkožní lipom, kollagenomy kůže a více obličejové angiofibrom (zejména horní ret). Tato funkce je někdy užitečné identifikovat takové pacienty a slouží jako příležitost k hlubšímu bádání. Se zvýšenou frekvencí F MEN karcinoidní tumory vyvíjí téměř výhradně primární lokalizovány v tlustém střevě a brzlíku, plic (bronchiální karcinoidu) a žaludeční sliznice. Zvýšená frekvence karcinoidů žaludku je pravděpodobně spojena s hypergastrinemic. Z nějakého důvodu, karcinoidní brzlíku je častější u mužů a karcinoid průdušky - u žen. Někdy se tyto nádory vylučují hormony (např ACTH) a často maligní. V některých případech pacienti s MEN 1 nalezeno myomu. Úmrtnost spojená s MEN 1, ostrůvek v důsledku rakoviny slinivky a maligní karcinoidní brzlíku.
Příčiny a patogeneze syndromu vícenásobné endokrinní neoplazie typu I (MEN 1)
MEN 1 je autosomálně dominantní rys. Vazba analýza ukazuje umístění defektního genu na dlouhém rameni chromozomu 11 (segment 11q13). Při analýze DNA z nádorové tkáně zjištěna ztráta alel v tomto lokusu (často v důsledku velké delece DNA). To vedlo k závěru, že gen, vada, která vede MEN 1, obvykle kóduje supresor proteinu růstu nádoru. Předpokládá se, že v přítomnosti normální alely na chromozomu dědí vadný druhá alela neaktivní, a teprve následné somatické mutace (často - vypuštění normální alely) určuje ztrátu funkce genu. Dominantní dědičnost v důsledku genetického defektu, zřejmě vysokofrekvenční takové delece. Ztráta aktivity genu, regulace buněčného růstu, vede k hyperplasii, což zvyšuje pravděpodobnost následných somatických mutací. Výsledkem je, že se buňky získají schopnost agresivnější a někdy maligní růst. Gen zodpovědný za MEN 1 (můži1), byli schopni identifikovat. Vady v tomto genu, kódující protein o 610 aminokyselinových zbytků (MužiV) patří nonsense mutace, missense mutace a delece. U pacientů s MEN 1 nalezeno asi 300 různých mutací genu, ale v 10-30% dostupných metod pro detekci jejich selhání. Produkt tohoto genu Menin, interakce se složkami histonu methyltransferázové komplexu, řídí expresi kinázy inhibitoru cyklin-dependentní p27Kip1 a p18Ink4c. Nedostatek Menin snižuje syntézu těchto inhibitorů, a tím dává na regulaci buněčného růstu. Kromě toho v nepřítomnosti syntézního Menin aktivuje transkripční faktor HLXB9, který také urychluje růst buněk.
V ojedinělých nádorů žláz s vnitřní sekrecí také často najít genové mutace můži1. Jeho defekt je detekován v buňkách 21% adenomů příštítných tělísek, gastrin 33%, 17% inzulínu, 50%, 36% vipom bronchiální karcinoidní nádory a 5% nádorů hypofýzy. Je důležité zdůraznit, že v 10-20% případů, i přes ztrátu heterozygotnosti na 11q13 lokusu, najít mutace můži1 v zárodečných buňkách nemůže. Na rozdíl od mužů 1I, charakter a selektivní destrukce jednotlivých endokrinních žláz nezávisí na specifických genových mutací.
Vyšetření syndrom vícenásobné endokrinní neoplazie I (MEN 1)
Klanicové MEN 1 méně než 1% všech nádorů hypofýzy, 2-4% případů primární hyperparatyreózy a asi 25% z gastrinu. Proto je genetická analýza ekonomicky odůvodněné pouze u pacientů s PPA. V nepřítomnosti MEN 1 rodinné historie můži1 zárodečné mutace zjištěných v 5% pacientů s gastrinomy, a u pacientů s 1-2% giperparati-reozom nebo prolaktinom. V těchto případech je genetická analýza by měla být provedena pouze u pacientů s vysokým rizikem (např, s odpovídajícími výrazy s příbuznými). Přeprava mutovaného genu je možno očekávat, že další rozvoj jiných endokrinních nádorů, což vyžaduje pečlivé sledování pacienta po dlouhou dobu. Na rozdíl od mužů 1I, můži1 detekce mutací by neměla vést k další intervence (např., Slinivky opakování). Takové zásahy nejsou lhostejní k pacientovi a jsou jen zřídka účinné. Nicméně, v případě, že pacient s hyperparatyreózy, kdy je operace hyperplazie příštítných tělísek, stejně jako v případě hyperparatyreózy relapsu, genetická analýza je nutné, a to i v nepřítomnosti jakéhokoliv označení MEN 1 v rodinné anamnéze. Při izolované tumorů hypofýzy (na rozdíl od PPA) pravděpodobnost MEN 1, jak je uvedeno výše, je velmi malý, a genetická analýza v tomto případě není nutné.
Ve všech případech je třeba vzít v úvahu historie dat rodiny. Pacienti z rodin s muži 1 úrovní by měla být kontrolována každoročně ionizované (nebo „upraveny“) vápníku a PTH. Úroveň gastrinu v séru také testovány ročně, a v přítomnosti PPA s příbuznými - ještě častěji. Další informace je možné získat za použití vzorku s sekretin. Poté, co příznaky hypoglykémie stanovení hladiny glukózy, inzulínu na lačno a proinzulinu. Stanovení pankreatický polypeptid (PP) může detekovat nádor neuroendokrinních buněk pankreatu. Ostrůvkových buněk nádory jiné vyhledávání jsou zobrazeny, snad jen v přítomnosti určitých příznaků a symptomů (dostatečným průjem, hypokalémie). Jak již bylo uvedeno výše, tyto nádory jsou obvykle tak malé, že je obtížné je detekovat i s nejmodernějšími metodami. To platí i pro nádory hypofýzy. V nepřítomnosti zjevných klinických projevů (známky zvýšené nebo snížené sekrece hormonů hypofýzy nebo projevy obklopují proces turecké sedlo) může omezit úroveň prolaktinu definice šarže, a pravděpodobně IGF-1 v séru. MRI hypofýzy a předním mediastinu a břišní CT provádí při léčbě a každé 3 roky.
Při sledování pacientů starších 10 let každoročně stanovení hladin gastrinu, vápník, albumin, PTH, půstu a PP. Někteří doporučují provádět každoroční CT nebo MRI břicha a hrudníku, a stanovení hladiny prolaktinu, a každé dva roky - provést MRI Sella. Jiní jsou omezena na provedení zobrazovací studia při manipulaci, a do budoucna - každé 3 roky.
Periodické studie by měly být po dobu alespoň 45 let, jako pravděpodobnost, MEN 1 v nosiči defektního genu, než tato doba je více než 95%. V dalších studiích se může provádět méně často. Je třeba mít na paměti, nicméně, že v pozdějším věku je riziko manifestace MEN 1 není snížen na nulu, a v některých případech jsou první projevy tohoto syndromu pozorován u jedinců, kteří daleko za 45. Po operaci nádoru (např paratyroidektomie) je nutná pokračovat v monitorování pacienta, nebude chybět opakování nebo na rozvoj dalších nemocí.
Léčba vícenásobné endokrinní neoplazie typu syndromu I (MEN 1)
Odstranění hyperparatyreózy v MEN 1 obvykle vyžaduje odstraňování tři a půl příštítných tělísek. Polovina čtvrtého žlázy je ponechána, aby se zabránilo rozvoji hypoparatyreózou. Také se používá celkové paratyroidektomie s příštítných plátky tkáně na opětovnou výsadbu na levém předloktí. V tomto případě zcela vyloučit možnost pooperační hypoparatyreoidismus stále nemůže, ale na druhou stranu, když mezisoučtu paratyroidektomie větší pravděpodobnost vzniku recidivující hyperparatyreózy. Během provozu profylakticky odstraní brzlík, se bát přítomnost v ní dalších příštítných tělísek nebo karcinoid.
Ukládání nebo recidivující hyperparatyroidizmus po chirurgickém zákroku u pacientů s MEN 1 jsou častější než v ojedinělých případech. Zvýšené hladiny hladiny vápníku a PTH v séru jsou uloženy po operaci u 38% pacientů s MEN 1, a zvýšená činnost příštítných tělísek relapsu (po 3 měsíce nebo více po redukci normalizaci hladiny kalcia) se vyskytuje u 16% z těchto pacientů. 8-12 let recidivy dosahuje 50%. Absence remise po chirurgickém zákroku je pravděpodobně kvůli přítomnosti přídavného příštítných tělísek (téměř 30% případů) nebo ektopické příštítných tělísek tkáně, jako recidiva spojené s zachování původní podnět k proliferace buněk příštitných tělísek. Opakované operace na krku při relapsu zvýšená činnost příštítných tělísek vedou k vyléčení u 90% pacientů, zatímco odstranění autograft - jen 60%.
Při gastrinomas, u pacientů s MEN 1 inhibitory používají hlavně protonové pumpy (omeprazol), potlačují sekreci kyseliny v žaludku. Konzervativní přístup je odůvodněno malou agresivitě takových nádorů velmi neúspěšné chirurgické postupy, stejně jako skutečnost, že nemocnost a úmrtnost pacientů je z velké části spojeny s hypersekrecí je kyselina chlorovodíková. Nicméně, mnoho gastrinomy (14%), jsou velmi agresivní a často jsou umístěny pod sliznice dvanácterníku a v lymfatických uzlinách kolem slinivky břišní. Chirurgická resekce nádoru s předběžného vytvoření jeho lokalizace v některých případech vést k pozitivním výsledkům. Vzhledem k zachování genetického defektu a multicentrické růst gastrinu možnost léčit většinu pacientů je velmi omezené, ale snížení hmotnosti nádorové tkáně může poskytnout dlouhé období nepřítomnosti příznaků. Symptomy hypergastrinémie Metastázy do jater a dalších orgánů oříznutí inhibitory protonové pumpy. V pokročilých případech, jako paliativní terapie pomocí systémovou chemoterapii a radiační terapii a selektivní embolizaci jaterních metastáz. Ostrůvkových buněk nádory pankreatu s průměrem větším než 3 cm, jsou často maligní a vyžadují resekci bez ohledu na jejich produkci hormonů.
Je důležité si uvědomit, že vápník stimuluje sekreci kyseliny chlorovodíkové. Odstranění hyperparatyreózy a hyperkalcemie často snižuje sekreci kyseliny a snižuje koncentraci v séru gastrinu. Paratyroidektomie obvykle normalizaci výsledků vzorku s sekretin. Přibližně 66% pacientů po těchto operací vyžaduje menší dávky H2-blokátory zmírnit příznaky PPA.
Naproti tomu, gastrin, insulinom zřídka lokalizované mimo slinivky břišní. Enukleace nádoru prostaty hlavy nebo slepé resekci těla a zadní části těla eliminovat hyperinzulinémie častěji než hypergastrinémie. Používají-li se kontraindikace zákroku (např., V přítomnosti těžkých průvodních onemocnění) nebo selhání identifikace nádoru lokalizační diazoxid nebo verapamil.
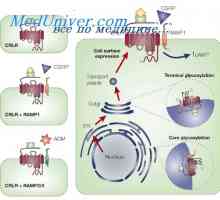 Primární hyperparatyreóza. Onemocnění kostí a hyperparathyroidismus
Primární hyperparatyreóza. Onemocnění kostí a hyperparathyroidismus Změny v ledvinách a kardiovaskulární systém s hyperparatyreózou
Změny v ledvinách a kardiovaskulární systém s hyperparatyreózou Diagnostika a léčba hyperparatyreózy
Diagnostika a léčba hyperparatyreózy Porucha příštítných tělísek (tetanie) morfologii, patologické anatomie
Porucha příštítných tělísek (tetanie) morfologii, patologické anatomie Forma příštítných tělísek adenomů morfologie, patologické anatomie
Forma příštítných tělísek adenomů morfologie, patologické anatomie Hyperparathyroid generalizované vláknitý osteodystrofie (von Recklinghausenova onemocnění)…
Hyperparathyroid generalizované vláknitý osteodystrofie (von Recklinghausenova onemocnění)…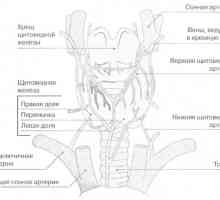 Štítné žlázy a příštítných tělísek
Štítné žlázy a příštítných tělísek Etiologie a patogeneze hyperkalcinemickou krizi
Etiologie a patogeneze hyperkalcinemickou krizi- Etiologie hypocalcemic krize
- Syndrom vícenásobné endokrinní neoplazie typu 1
- Hyperparatyreóza (generalizovaná fibrózní osteodystrofie, Recklinghausen nemoc) -zabolevanie v…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Hyperparatyreóza: účinný provoz tablety
- Primární hyperparatyreóza je spojena s fibromyalgie?
 Syndromem mnohočetné endokrinní neoplazie, typ II (Maine 2): příčiny, příznaky, léčba, symptomy
Syndromem mnohočetné endokrinní neoplazie, typ II (Maine 2): příčiny, příznaky, léčba, symptomy Vláknitý osteodystrofie hyperparathyroid
Vláknitý osteodystrofie hyperparathyroid Renální osteodystrofie
Renální osteodystrofie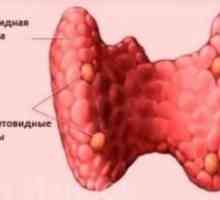 Sekundární hyperparatyreóza: léčba, příznaky, příčiny, příznaky
Sekundární hyperparatyreóza: léčba, příznaky, příčiny, příznaky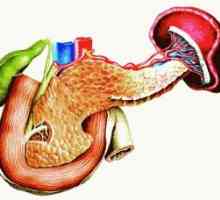 Dědičné pankreatitida: příznaky, léčba, příznaky, příčiny
Dědičné pankreatitida: příznaky, léčba, příznaky, příčiny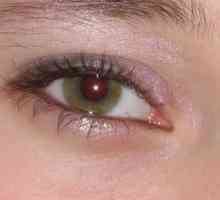 Zrakové postižení v onemocnění příštítných tělísek
Zrakové postižení v onemocnění příštítných tělísek Primární hyperparatyreóza, příznaky, léčba, příčiny, příznaky
Primární hyperparatyreóza, příznaky, léčba, příčiny, příznaky