Změny v orgánu vidění v vrozené patologii traumatické oblasti obličeje

Tělo se mění ve vizi s vrozenými vadami kraniofaciální oblasti, s přihlédnutím k umístění orgánu vize, lámání formu lebky (zejména oblasti obličeje) vedou k závažným změnám v různých částech vizuálního analyzátoru.
Syndrom charakterizovaný kraniosynostózou
V těchto onemocnění jsme pozorovali předčasné fúzní svary, což vede k deformaci lebky. Tato skupina dědičných syndromů zahrnují turricephaly, crouzonův syndrom, Apertův, Pfeiffer, Setre-Hotz, Carpenter.
turricephaly
Klinické příznaky. Když tento syndrom generovaný specifický tvar lebky - věž lebky. Taková deformace lebky se vyvíjí v důsledku předčasného fúzi lebky stehy, a zejména koronální stehem. Ve většině případů, zvýšeného nitrolebního tlaku.
oční příznaky. U dětí, existuje významný pokles vidění, což je dáno jednak s vrozenou oční atrofie, stejně jako s tupozrakosti, šilhání dojde v důsledku. Vyznačující se tím, že sníží velikost očnice a proptosis. Pozorovaná nystagmus.
Lebka jako listů jetele (syn. Trojlístek)
V okamžiku, kdy dojde k dědičnost neizvesten- se stejnou frekvencí u obou pohlaví.
Klinické příznaky a symptomy. Charakterizované rozvojem deformace lebky čtyřlístku nebo trojlístku - lebka je tupý, trilobed tvar, vysoké čelo, zobákovitý nos, střední část obličeje hypoplazie. Změny v lebce způsobené nitroděložním fúzí koronálních a lamblovidnogo švech. Tam je hydrocefalus. Patologie lebky v kombinaci s abnormalitami páteře, ankylózou loketních a kolenních kloubů, syndaktylie rukou a nohou, poruchy dýchacího traktu. Děti s tímto syndromem umírá v mladém věku.
Oční příznaky. K dispozici je výraznější exophthalmos, což je vzhledem k prudkému poklesu objemu oběžné dráhy. Charakteristické dislokační bulvy. Nesmykanie oční štěrbiny vést ke vzniku keratitidy. Charakteristika antimongoloidny sekční oční štěrbiny a ptózy.
Crouzonův syndrom, kraniofaciální Dysostosis
Rodina a dědičné onemocnění charakterizované abnormalitami mozkové nebo obličeje lebky. Typ dědičnosti - autozomálně dominantní. Je zde možnost sporadické výskytu choroby. Etiologie a patogeneze nejsou zřejmé. Podle stejného onemocnění často postihuje muže i ženy.
Klinické příznaky a symptomy. Dědičné synostózy z lebky (předčasné osifikace zejména koronární a lambdoid stehy) za vzniku typickou konfiguraci a charakteristické rysy obličeje: acrocephalic expanzi lebky, „orlím“ nosu. Subulate zuby jsou vzácné, velké pero, vysoká patra, rozštěp patra a uvula je zvukovod atrezie, hluchota. Když X-ray může odhalit typické znaky: porézní strukturu lebky, připomínající plástve, uzuratsii. Kombinace s syndaktylie rukou a nohou.
Oční příznaky. Vzhledem ke snížení objemu drah pozorovaných exophthalmos, a v některých případech, a dislokace oční bulvy. Zvýšený nitrolební tlak a snížení velikosti zrakového nervu kanálů vedou k rozvoji městnavého struků s následným zrakového nervu atrofie. Vyznačující se tím, hypertelorismem, rozbíhající strabismu, nystagmus, Mongoloid tvaru oka, modré skléry. V některých případech, šedý zákal. Vzhledem k výše popsané změny jsou již snížena zraková ostrost v raném dětství.
} {Modul direkt4
Apertův syndrom
Dědičné onemocnění, na základě vývoje, z nichž je prenatální poškození první žábry oblouk s rozvojem poruchou embrya.
Klinické příznaky a symptomy. Tvořena typickou obličeje anomálie připomínající ty s crouzonův syndrom. Rozvíjení hydrocefalus. Změny v lebce v kombinaci s tvorbou tracheo jícnu píštěle a jiné vady gastrointestinálního traktu, vrozených srdečních vad, abnormalitami páteře. Tam je mentální retardace.
Oční příznaky. Vzhledem ke snížení objemu očních důlků tam exophthalmos. Pozorovaná hypertelorismus, antimongoloidny oči, ptóza, exotropia. Rozvoj městnavé disk následovaný atrofie očního nervu. Charakterizovány výskytem keratokonu, ektopie části čočky, katarakta, pigmentového degenerace sítnice. Možná, že vývoj vrozených glaukomu.
Pfeiffer syndrom
Dědičné onemocnění, které je založeno na předčasné synostózy koronárních a lambdoid stehy.
Klinické příznaky. Je-li tento syndrom tvořena deformace lebky akromegalie typu, výjimečně tvar lebky podobá jetel. Charakteristické rysy obličeje jsou následující: zaostalé krátký rovný nos, hypoplazie maxily, mandibuly prognanizm. Utero rozvíjet hydrocefalus. Změny lebka v kombinaci s mírnými Syndaktylie prsty II-III a II-IV prsty. Charakteristickým rysem je přítomnost širokých palec a prsty a končetiny varozity deformity. Možná, že při selhání ledvin anomálií, choanal, srdečních vad. Mentální retardace je méně častá než v Apertův syndromu.
Oční příznaky. Charakterizované rozvojem epicanthus, šilhání. V některých případech, poznamenal vzhled coloboma duhovky a afakie.
Syndrom Setre-Hotz
Tento syndrom je označován akrotsefalosindaktelii typově III se dědí autosomálně dominantním způsobem.
Klinické příznaky. Vzhledem k nerovnoměrnému vytvořen kraniosynostózou lebeční asymetrie. Typické změny zahrnují obličeje: zobákovitý nos, se odchýlil septum, nízkou úroveň růstu vlasů na čelo, uši dolní polohy s charakteristickými příliš zakřivené spirály. Existuje vysoká nebe. Změny v lebce v kombinaci s kožní syndaktylie prstů na rukou a nohou - zpravidla, prsty 11-III. Vyznačující se tím, zkrácené formy prstů.
oční příznaky. Tam hypertelorismus kombinaci s antimongoloidnym sekčními oční štěrbiny a ptózy. K dispozici je cross-eyed, a v některých případech i optická atrofie. V důsledku zúžení slezonosovogo kanál může zhoršit slzy odtok. Vzhled exoftalmem není typické.
Carpenter syndrom
Tento syndrom se označuje akrotsefalosindaktelii typově II dědičné autozomálně recesivní způsobem.
Klinické příznaky. Vzhledem k tomu, předčasné synostózy vytvořeného turricephaly a specifický tvar obličeje - plochý hřbet nosu, lícních velké, nízké umístění boltce. Změny v lebce v kombinaci s abnormalitami prstech rukou a nohou - syndactyly, clinodactyly, zkrácení střední falangy, zdvojnásobení palce, stejně jako s valgus deformity kolenního a varus nohy. Porucha je často doprovázena mentální retardace, hypoplazie pohlavních orgánů, obezity, srdečních chorob.
Oční příznaky. Charakterizuje vývoj hypertelorismem, epicanthus a telekantusa.
Syndromy charakterizované tvorbou trhlin
Vývoj této skupiny syndromů spojených s narušenou složených sousedními útvary během embryonálního vývoje.
Goldenara syndrom (syn. Oculo-ušní-vertebrální dysplazie)
Kongenitální onemocnění charakterizované abnormalitami složité - zejména očí, uší a míše. Typ dědičnosti pravděpodobně autozomálně recesivní.
Klinické příznaky. Často atrézie zevního zvukovodu, středního ucha abnormality a hluchota. Tvarové anomálie žeber a obratlů. Tam je mentální retardace.
Oční příznaky. Vyznačující se tím, spojivek patologie jako epibulbar dermoid lipodermoidov nebo bílé nebo nažloutlé barvy s hladkým povrchem, obvykle umístěné v dolní vnější části oční bulvy, colobomas horního víčka (zřídka nižší) oční. Mnohem méně často pozorovány mikroftalmie, microcornea, duhovky coloboma, ptóza, strabismu, šedý zákal. Tyto změny ve většině případů jednostranné.
Treacher Collins syndrom,
Dědičnost onemocnění souvisejících s genem se nachází na dlouhém rameni chromozomu 5. Tam je autosomálně dominantní vzor dědičnosti.
Klinické příznaky a symptomy. Typické obličejové změny - hypoplazie lícní kosti a dolní čelist, nos zploštění a posunutí dolní čelisti zpět, zhoršená tvorba chrupu. Hoan dojít k atrofii, vývojové abnormality vnějšího ucha, hluchota.
Oční příznaky. Tam jsou změny ve tvaru víčka (coloboma vnější třetině dolního víčka) a očního koutku - antimongoloidny řez. Změna tvaru věku vedou k rozvoji degenerativních procesů v koutku oka. Tam je obstrukce naso-slzných kanálků. Objeví dermoid cysty, lokalizované na limbu a na oběžné dráze.
 Apertův syndromu u plodu. Diagnostika a prognózu Apertův syndromu
Apertův syndromu u plodu. Diagnostika a prognózu Apertův syndromu Pfeiffer syndrom. Diagnostika a prognóza Pfeiffer syndromu
Pfeiffer syndrom. Diagnostika a prognóza Pfeiffer syndromu Tanatofornaya dysplazie. Diagnózu a prognózu tanatoformnoy dysplazie
Tanatofornaya dysplazie. Diagnózu a prognózu tanatoformnoy dysplazie Embryo lebka. Tvorba fetální lebky
Embryo lebka. Tvorba fetální lebky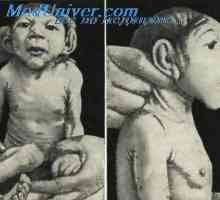 Kranioskhizis a fetální mikrocefalie. encefalokéla embryo
Kranioskhizis a fetální mikrocefalie. encefalokéla embryo Fetální lebky. Tvorba lebky embrya
Fetální lebky. Tvorba lebky embrya Osifikace fetální lebky. Tvorba lebky embrya
Osifikace fetální lebky. Tvorba lebky embrya Věkové rozdíly ve struktuře lebky
Věkové rozdíly ve struktuře lebky Zlomeniny kostí lebky
Zlomeniny kostí lebky- Studie parametry baze lební u pacientů s horní čelisti retropolozheniem
- Kosti lebky, kromě dolní čelisti jsou propojeny nepřetržité spojení. Sklíčka kosti lebky jsou…
- Kostra hlava je lebka, lebka, některé kosti, které jsou rozděleny do kosti lebky. Ossa cranii a…
- Jedním z rysů lebky novorozence jsou fontanelles, fonticuli jako celek. Představují neokostenevshie…
- Hydrocefalus-nárůst v mozkomíšním moku v lebeční dutině. Etiologie, patogeneze. Porušení resorpce a…
- Exophthalmos vystoyanie oční bulvy vpředu. Etiologie, patogeneze: další tkáně očnice, v první řadě…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Výborná lékařská encyklopedie IC nevronet. choroba
- Výborná lékařská encyklopedie IC nevronet. choroba
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Vědci: proč datle neporušují lebku
 Hypertenzí Hydrocefalus syndrom u dětí, příčiny, příznaky, léčba
Hypertenzí Hydrocefalus syndrom u dětí, příčiny, příznaky, léčba