Dědičné choroby nervového systému, svalová dystrofie, myasthenia gravis
Video: ekzom pro neuromuskulárních onemocnění, DV. Vlodavets, Výzkumné centrum pro genetické medicíny

Progresivní degenerativní onemocnění nervového systému
Progresivní degenerativní onemocnění nervového systému, výsledkem z geneticky dané vady nebo patologie vývoje embrya. Běžné projevy těchto onemocnění jsou: degenerativní charakter a soudržnost nervové tkáně, progresivní průběh. Mezi ně patří, zejména, syringomyelie, v nichž mícha tvoří dlouhý dutý prostor, ničit zadního rohy. To vede k defektu bolesti a teploty citlivost, Atropatena.
Skupina dědičné ataxie poměrně četné, jejich hlavním projevem ataxie je spojena s patologií mozečku traktu nebo hluboké citlivosti.
Amyotrofická laterální skleróza (ALS) - těžké a rychle postupující onemocnění, Sopron dráhy v míše. Proto je kombinace atrofické parézy a pyramidálních příznaků, to znamená, že rozvojové pohybové poruchy jsou charakterizovány jak v periferní a centrální obrny.
Parkinsonova choroba je progresivní onemocnění, které je založeno na pigmentovou primární lézí dopaminergních neuronů substantia nigra hustou část a pigment jiné kmeni. Riziko pro další fázi pacienta příbuzný je asi 10 krát vyšší než v populaci. Pro Parkinsonova choroba je charakterizována triádou příznaků: třes, zvýšení svalového tonu a gipokineziya- kritéria diagnózy jsou velmi složité. klinika onemocnění se vyskytuje pouze v případě, zabila více než 80% neuronů. V některých případech se nemoc svůj debut ve věku 18 let (tzv juvenilní parkinsonismus), z větší části - v pozdějším věku. To klade vysoké nároky na aktuálnost a adekvátnost léčby.
Moderní adekvátní terapie poskytuje mnoho příležitostí k dobré kompenzaci narušených funkcí, jakož i zachování či obnovení sociální adaptace.
Dědičné choroby nervosvalové
Degenerativní onemocnění nervosvalové - onemocnění postihující převážně neuromuskulární systém dědičné povahy. Oni jsou také označovány jako progresivní neuromuskulární dystrofie (PMD), a představují nejvýznamnější skupinu ze všech dědičných chorob.
Klasifikace progresivní svalovou dystrofií
- Progresivní svalová dystrofie - geneticky určená porucha s primární progresivní degenerativní změny ve svalech (není primární patologii periferního motoneuronu). Ve svalové tkáně, která je cílem defektu primární genu, kvůli které je abnormální syntézu svalového proteinu myodistrofina a jeho rozklad je urychlen. Porážka nervového systému s myopatií je sekundární.
- Spinalkye amyotrofie - geneticky podmíněné primární léze míchy předních rohů se sekundární progresivní ochablé a svalové atrofie.
- Neural amyotrofie - primární geneticky určená syndrom polyneuropatie (což mielinopatii) s vývojem sekundární amyotrofie senzorických a autonomním abnormalit.
Primární progresivní svalová dystrofie
Různé formy onemocnění se vyskytují v různých věkových kategorií - od 1-2 do 40-50 let věku a starších. Jsou charakterizovány motorovým nemotornost, nestability, pády při chůzi, únavu. Dítě se objeví strach a neochotu chodit. U pacientů s chůzí tvořil je „kachna cesta“ - kolébala.
Pro některé formy charakteristické psevdogipertrofiya svalů, často postihuje lýtkové svaly: oni atrofii s maskovací atrofie a dokonce zvětšovat v důsledku šíření pojivové, tukové tkáně. Slabost a atrofie svalů nejprve lokalizovaných v svalů pánevního pletence s maximální exprese v proximálním nohy.
Existuje výrazná bederní lordóza, skolióza, „křídla“ čepele, úzký „vosí“ pas. Zvedání ze sedu je obtížná a děti uchýlit k pomocným zařízením (Gowers techniky), -. „Lezení po žebříku“, „lezení na sebe“ Existují případy, kdy s rozvojem demence. Trpí srdeční sval. Dále pacienti ztrácejí schopnost chodit samostatně. Tento způsob zahrnuje kardiovaskulární systém (vyvinutý dilatační nebo hypertrofická kardiomyopatie).
Sekundární - spinální a neurální svalová dystrofie
Spinální svalová dystrofie (amyotrofie) se dědí autosomálně recesivním způsobem. Gene sleep-ních svalová atrofie mapovány na chromozomu 5gl1.2-13.3.
Může odhalit časné příznaky onemocnění tabloid. Opožděný vývoj motoru. Během elektromyografie detekovány léze předních rohů míchy. k progresi onemocnění.
Při spinální amyotrofie formy na výměře samotných potenciálů elektřiny-ogramme zaznamenán fibrillyatsiy- rychlost končetiny nervových vzruchu relativně konzervované, ale může snížit v důsledku úmrtí míšních motorických neuronů.
Nejčastějším z nich je neuronová amyotrophy neurální amyotrophy Charcot-Marie-Tooth choroba. Je to v jejich klinických projevů připomínající senzorimotorické polyneuropatie, distálně zvýrazněna a počínaje chodidel a nohou. Proudí benigní, pomalu. V průběhu času se tvořily charakteristickou deformaci chodidel - typu „čáp nohy“ nebo „kalhoty“: tenký v důsledku atrofie svalů dolní končetiny s neporušenými boky. Za prvé, „pád“ Achilles reflexy, pak redukuje koleno.
Když elektronejromiografii zaznamenán pokles hrubé končetiny rychlost šíření nervový impuls.
Myasthenia gravis. Myastenický a cholinergní krize
myasthenia (Miastenia gravis pseudoparalitica) - závažné neuromuskulární autoimunitní onemocnění charakterizované abnormální únavy a slabosti příčně pruhovaného svalstva (Akimov GA je stejná MM, 2000).
etiopatogeneze. Hlavní spojení - výskyt autoprotilátek proti nikotinové holinoretseptorami koncové desce svalových vláken a blokovat neuromuskulární přenos. K dispozici je odkaz na patogenezi myasthenia gravis prostaty léze brýlové výtah. Thymom často detekována (až 40% případů), zřídka - atrofie thymu.
klinika. Myasthenia gravis může nastat v jakémkoli věku, ale častěji - mezi 16 a 40 let, ale jsou zde dříve a později formy (označené výskyt píky při 30 až 70 let). Ženy trpí častěji než muži. Hlavním příznakem - abnormální svalová únava s rozvojem jejich rostoucí slabost v opakovaných pohybů, jako je například výskyt duchů nebo ptózy ve čtení.
Rozvoj svalové slabosti v myasthenia se liší od periferní nebo centrální parézou v tom, že opakování pohybů, a to zejména v rychlým tempem, prudce zvyšuje a může dosáhnout plný rozsah paralýzy. Po odpočinku, spánku, první pohyby mohou být normální, ale při následné objeví únava, míra, která postupuje s pokračujícím zatížení.
Myastenický epizoda se může vyvinout u dětí, které se narodily matkám trpících myasthenia gravis (tzv myasthenia gravis novorozenců). Míra pohybových poruch odškodnění může být zcela dostačující (pro samoobslužný provoz v domácnosti), špatná (vyžaduje péči outsider). Nejvážnější komplikace myasthenia - krize myasthenic.
krize myasthenic - vytvoření náhlé havarijní kritického stavu v důsledku blok nervosvalového vedení. Mezi hlavní příznaky - rychle rostoucí, zobecněné svalová slabost, dosahující do tetraplegii úrovni.
komplikace:
- respirační poruchy během formě bulbární
- riziko obstrukce dýchacích cest hromadí husté sputum,
- možnost vdechnutí potravy nebo „udušení ventilu“ kvůli jazykovým depresí a slabých stránek epiglottis,
- off bránice a mezižeberní slabosti dýchacích svalů.
Video: Toto video je o celou pravdu DMD, nemoci, která nešetří jeden
Předávkování anticholinesterázy léků může vést k cholinergní krizi prudkému zhoršení zdraví. Pomoc při mimořádných událostech „je zdravotničtí pracovníci na jednotce intenzivní péče nebo jednotky (oddělení) na jednotce intenzivní péče.
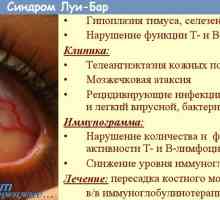 Imunodeficience syndrom ataxie-teleangiektázie. Louis-Bar syndrom
Imunodeficience syndrom ataxie-teleangiektázie. Louis-Bar syndrom První pomoc při nerovnoměrného asymetrické více lézí
První pomoc při nerovnoměrného asymetrické více lézí- Bolesti hlavy pro pomalé infekce
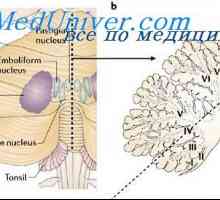 Mozečku. Projevy patologie mozečku
Mozečku. Projevy patologie mozečku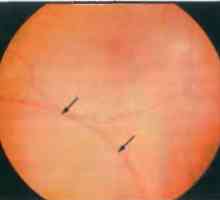 Nemoci obvodu sítnice: degenerativní retinoschisis
Nemoci obvodu sítnice: degenerativní retinoschisis- Peroxisomovým nemoc
- Bezbolestné oftalmoplegie
- Myasthenia gravis. Historie vývoje, patogeneze
- Střevní malabsorpce (sprue) -simptomokompleks, poruchy vyplývající z vstřebávání v tenkém střevě.…
- Amyotrofická laterální skleróza (neuronová onemocnění motoru) spastikoatroficheskie trvale…
- Progresivní svalová dystrofie podstatné progresivní degenerace svalové tkáně, plynoucí z jakéhokoli…
- Syringomyelie, chronické onemocnění charakterizované tvorbou dutin v míše a medulla oblongata k…
- Spinální amyotrofie skupina dědičné chronické onemocnění charakterizované progresivní atrofické…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Seznam nemocí velká lékařská encyklopedie
 Demyelinizační onemocnění nervového systému: příznaky, léčba, příčiny
Demyelinizační onemocnění nervového systému: příznaky, léčba, příčiny Mozečku Poruchy: příčiny, příznaky, příznaky, léčba
Mozečku Poruchy: příčiny, příznaky, příznaky, léčba Goniodisgenez
Goniodisgenez