Genetické poruchy, které vedou k neplodnosti u lidí

Video: genetická stabilita a zničení bílé rasy. Cizí geny a důsledky
Na molekulární úrovni, příčiny neplodnosti u lidí byly málo studována.
Většina známých mutace vedou k nedostatku nebo zpoždění puberty a v důsledku toho, neplodnosti. Ale doktor o neplodnosti léčit lidi, kteří mají pohlavní vývoj je normální. Screening u většiny mutací, které vedou k neplodnosti, praktický smysl teď nezáleží. Nicméně, některé případy si zaslouží zvláštní zmínku, protože se často setkáváme v každodenní praxi.
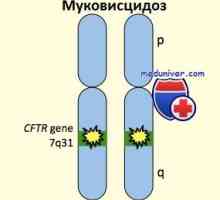
Bilaterální aplazie chámovodu
Bilaterální aplazie chámovodu je přítomen v 1-2% neplodných mužů. Podle většiny údajů v 75% případů se nacházejí, vyznačující se tím, že mutace v genu pro CF vyplývající u cystické fibrózy. Hlavní riziko v těchto případech - možnost narození dítěte s cystickou fibrózou. By měly být vyšetřeny na přítomnost mutací obou partnerů, a pak podržte příslušné poradenství. Jsou-li oba partneři jsou nositeli cystickou fibrózou, hrozí, kdy dítě dosáhne 25% (v závislosti na povaze mutace). Dokonce i když člověk našel jen jednu mutaci vedoucí k cystické fibróze, a nositel ženě není, je lepší hrát na jistotu a poslat pár konzultovat genetika. Přibližně 20% z dvoustranné aplazie chámovodu je doprovázeno vývojem vady ledvin a v jedné studii, tito pacienti neodhalila mutace vede u cystické fibrózy (i když se počet analyzovaných mutace byla malá).
Je třeba zdůraznit, že účelem screeningu je odhalit spíše než cystická fibróza aplázie. Kombinace mutací způsobujících Aplazie chámovodů, různorodá a složitá, takže je obtížné poradenství v této nemoci. První studie o genetice bilaterální aplazie chámovodu, nebyl jeden člověk homozygotní pro AF508 mutací, nejčastější mutací v genu CF, což je klasická forma cystické fibrózy se vyskytuje v 60-70% případů. Přibližně 20% pacientů jsou jen dvě mutace v charakteristice genu CF cystické fibrózy, - v mnoha případech to missensmutatsii (kombinace dvou alel, což způsobuje mírné cystickou fibrózu, nebo jednu alelu způsobující mírnou formu onemocnění, a jeden - těžké). Také nalezený polymorfismus v intronu 8, kde počet různých alel na thymin je 5, 7 nebo 9. V přítomnosti alely 5T během transkripce vynechán exon 9 a mRNA a protein v budoucnu a zkracuje. Nejběžnější genotyp s oboustranným aplazie vas deferens (o 30%) - kombinace alel nesoucích mutaci způsobující cystickou fibrózou, a 5T alely.
Mutace R117H zahrnuty v kontrole hmotnosti, protože jeho kombinace s dalšími těžšími mutací genu CF může způsobit cystickou fibrózou. Když R117H detekce mutací chování odvozených testování přítomnosti polymorfismu 5T / 7T / 9T. Po detekci alely 5T je třeba zjistit, zda je na jednom chromozomu R117H (m. E. cis) nebo různé (trans). 5T alela v „pozici s ohledem na R117N způsobuje cystickou fibrózu, a pokud žena je také nositelem jedné z alel, které způsobují onemocnění, riziko cystické fibrózy u dítěte je 25%. Složitost genetiky cystickou fibrózou je zřejmé, když se podíváte na rozmanitosti fenotypů v homozygotů pro alely 5T. Přítomnost alely 5T snižuje stabilitu mRNA, a je známo, že pacienti s nemodifikovaným úrovni mRNA 1-3% normálu, cystická fibróza vyvíjí v klasické formě. Když je úroveň intaktní mRNA je větší než 8 až 12% normálu, nemoc neprojevuje, a v mezilehlých úrovních různé varianty jsou možné, a to od bez symptomů onemocnění až do bilaterální aplazie chámovodu a jemným cystickou fibrózou. Je třeba také poznamenat, že aplazie chámovodu v mírných případech je i jednostranné. U běžné populace alela 5T dochází s frekvencí asi 5%, s jednostranným aplazie chámovodu - na 25% a na bilaterální aplazie - s frekvencí 40%.
American College lékařské genetiky a American College of porodníků a gynekologů doporučit detekovat pouze 25 mutace výskyt v populaci USA, která není menší než 0,1%, a analýza polymorfismu 5T / 7T / 9T provádí pouze jako derivát testu. V praxi se však mnoho laboratoří může snížit náklady tím, že umožňuje analýzu hlavního programu, který, jak je uvedeno výše, může vést k obrovským potížím při interpretaci výsledků. Je třeba připomenout, že účelem prověření - identifikace cystickou fibrózou.
Geny regulující spermatogeneze
Geny pravděpodobně odpovědné za spermatogenezi, Y mapované na xpomosome v AZF oblasti nacházející se na určitém místě, Yq11 (U SR gen umístěn na krátkém raménku chromozomu Y). Ve směru od centromery k distální části ramenních částí uspořádány postupně AZFa, AZFb a AZFc. AZFa na pozemku jsou gen a USP9Y DBY, na místě AZFb - gen komplexní RBMY a plocha / 4Z / c - DAZ genu.
Některé z genů, podílejících se na regulaci spermatogeneze, genom je zastoupena ve více kopiích. Zdá se, že v genomu má 4-6 kopií genu DAZ a 20-50 geny nebo pseudogeny RBMY rodiny. DBY USP9Y a prezentovány v genomu jedné kopii. Vzhledem k velkému počtu opakovaných sekvencí a rozdíly v designu studie analýzy Y-chromozomálních oblastech ovládajících spermatogenezi, s sebou nese problémy. Například detekce delecí v AZF provádí většinou pomocí analýzy DNA markerové místa, krátké sekvence DNA, známého chromozomální lokalizace. Čím více se analyzuje, tím vyšší je pravděpodobnost pro detekci delece. Obecně platí, že delece v oblasti AZF poháru našel u neplodných mužů, ale existují případy, detekce a zdravé.
Důkaz, že AZF oblast obsahuje geny, které regulují tvorbu spermií sloužil USP9Y intragenní deleci v genu, který je také nazýván DFFRY (jako odpovídající homologní genu Drosophila FAF). U neplodných mužů byla nalezena delece čtyř párů bází, což nebylo jeho zdravý bratr. Tato pozorování, spolu s analýzou dat in vitro naznačují, že mutace v genu USP9Y porušuje spermatogeneze. Opakovaná analýza dříve publikovaných dat, výzkumníci zjištěn další deleci v jednom genu USP9Y porušuje spermatogeneze.
Přehled průzkumu dat v blízkosti 5000 neplodných mužů k mutací v Y-chromozomu ukázalo, že přibližně 8,2% času (ve srovnání s 0,4% u zdravých) jsou delece v jedné nebo více oblastech AZF oblasti. V některých studiích čísla v rozmezí od 1 do 35%. Podle výše uvedeného průzkumu, nejčastější místo delece v AZFc (60%), poté - v AZFb (16%) a AZFa (5%). Další případy - kombinace delece ve více místech (často včetně delecemi v AZFc). Většina mutací byly nalezeny u mužů s azoospermií (84%) nebo se závažnou oligozoospermie (14%) je definována jako počet spermií nižší než 5 milionů / ml. Interpretace dat delecí v AZF je velmi obtížné, protože:
- oba jsou neplodné a zdravých mužů;
- Přítomnost DAZ clusteru a RBMY, obsahující více kopií genu, komplikuje analýzu;
- různé studie zkoumaly různé parametry spermatu;
- sada Y chromozomu kontigovyh karty v důsledku přítomnosti repetitivních sekvencí nebyla dokončena;
- To nebylo k dispozici dostatek údajů u zdravých mužů.
Ve dvojitě zaslepené studii u 138 mužů, z páry, kteří hledají lékaře pro IVF, 100 zdravých mužů a 107 mladých dánských vojáků byly zjištěny hladiny pohlavních hormonů, parametry spermatu a držel analýzy regionu AZF. Pro studii bylo použito AZF oblast 21 DNA místo označení v normálních parametrů spermií, a ve všech případech, kdy počet spermií přesáhla 1 mil / ml, bylo zjištěno, delece. V 17% případů idiopatické azoospermie nebo kriptozoospermii a 7% s jinými typy azoospermie a kriptozoospermii identifikována delece část AZFc. Je zajímavé, že žádný z účastníků studie bylo zjištěno, delecí v oblastech AZFa a AZFb. To naznačuje, že geny se nachází v oblasti AZFc, nejdůležitější pro spermatogeneze. Později větší Studie byla provedena, které byly získány obdobné výsledky.
Při identifikaci delece v chromozomu Y by měla projednat s oběma potenciálními rodiči. Hlavním rizikem pro potomstvo je, že synové mohou dědit toto odstranění od otce a jsou neplodné - tyto případy jsou popsány. Účinnost IVF a míra těhotenství těchto delecí, se zdají mít žádný vliv.
Fragilního X chromozomu u žen s předčasným selháním vaječníků
V ojedinělých případech, předčasné ovariální selhání v asi 2-3% žen vykazují přítomnost premutace v FMR1 gen zodpovědný za vznik hromosomy- syndrom fragilního X chromozomu u žen s předčasným selháním vaječníků dědičné frekvence tohoto premutace dosahuje 12-15%. Křehký úsek v Xq28 lokusu může být identifikován Karyotyping buňky kultivovány za podmínek nedostatkem folátu, ale to se obvykle provádí analýzu DNA. Syndrom nebo Fragile-X se týká nemocí, které jsou způsobeny rostoucí počet trinukleotidových opakování: normální FMR1 gen obsahuje méně než 50 opakujících se TSTSG sekvence nosiče číslo premutace je 50 až 200, zatímco muži se syndromem nebo Fragile-X - 200 ( plné mutace). křehký syndrom X chromozomu charakterizován X-spojený dominantní dědičnosti s neúplnou penetrací.
Identifikovat nosiče premutace důležité, protože mohou být, a dalšími členy rodiny: mohou být narozené syny s fragilního X chromozomu, což samo o sobě projevuje mentální retardace, charakteristické rysy obličeje a makroorhizmom.
Sekundární hypogonadismus a Kallman syndromu u mužů
U mužů se syndromem, vyznačující se tím, Kalman anosmií a sekundární gipogonadizm- také možné obličeje deformit střední čáry, jednostranné ageneze ledvin, a neurologických poruch - synkineses, oculomotor a cerebelární poruchy. Kallman syndrom je charakterizován X-vázanou recesivní způsob dědičnosti a je způsobena mutacemi v genu KALI- naznačují, že Kalmanův syndrom způsobilo 10-15% případů izolovanou deficiencí gonadotropinů u mužů s anosmií. Nově objevené autosomálně dominantní formu Kalmanova syndromu, což způsobuje mutace v genu FGFR1. V případě, že izolovaný deficit gonadotropinů bez anosmií často vyskytují mutace v genu s receptorem GnRH (gonadotropin uvolňujícího hormonu gen receptoru). Nicméně, oni tvoří jen 5-10% všech případů.
Video: Dědičná diagnóza lidská nemoc a prevence. umělé oplodnění
 Vědci našli nový způsob, jak se vyrovnat s neplodností
Vědci našli nový způsob, jak se vyrovnat s neplodností- Nízká porodní váha - neplodnost v budoucnosti?
- Stres a neplodnost u mužů
- Neúspěšná léčba neplodnosti a emocionální zdraví žen
 Žloutenka u dítěte s cystickou fibrózou
Žloutenka u dítěte s cystickou fibrózou Mutace gonadotropin geny. Mutace v podjednotky LH a FSH
Mutace gonadotropin geny. Mutace v podjednotky LH a FSH Mutace gonadotropin receptory. Abnormality LH a FSH receptoru
Mutace gonadotropin receptory. Abnormality LH a FSH receptoru Cysta-stop-genitální syndrom. Genetika mužské neplodnosti
Cysta-stop-genitální syndrom. Genetika mužské neplodnosti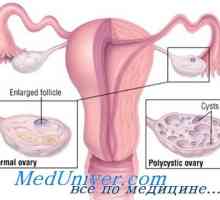 Předčasné selhání vaječníků, Příčiny předčasného neplodnosti
Předčasné selhání vaječníků, Příčiny předčasného neplodnosti- Proteiny imunitního systému způsobit mutace DNA, které vedou k rakovině
- Vědci našli genetickou příčinu akutní lymfoblastickou leukémií
- Nalezeno léčení vrozených onemocnění očí
- Jaké jsou genetické choroby
 Gene plodu onemocnění. Prenatální diagnóza fetálních genetických chorob.
Gene plodu onemocnění. Prenatální diagnóza fetálních genetických chorob. Biochemické prenatální diagnóza cystické fibrózy, adrenální hyperplazie.
Biochemické prenatální diagnóza cystické fibrózy, adrenální hyperplazie. Léčba mužské a ženské neplodnosti kliniky Tambre Španělsko, Madrid
Léčba mužské a ženské neplodnosti kliniky Tambre Španělsko, Madrid- Onkologiya-
- Nový typ antikoncepce
- Nová kombinace léčiv prodloužit životy lidí s cystickou fibrózou
 Cystická fibróza: symptomy, léčba, diagnostika, příznaky, příčiny
Cystická fibróza: symptomy, léčba, diagnostika, příznaky, příčiny Mutace, které vedou k dědičných chorob u lidí
Mutace, které vedou k dědičných chorob u lidí