Medulární karcinom štítné žlázy

Video: medulární a dobře diferencovaný karcinom štítné žlázy: inovativní prognostické, prediktivní a terapeutické přístupy
Na MTC představuje 5-10% všech zhoubných nádorů štítné žlázy.
Přibližně 75% z MTC je sporadická, a zbytek - rodina spojena s jedním ze tří genetickými syndromy: rodina izolované MTC, vícenásobné endokrinní neoplazie na [MEN PA (MTC, feochromocytomu a primární hyperparatyreózou)] nebo MAN PB [MTC, feochromocytom , více slizniční neuromy a (zřídka) primární hyperparatyreóza].
V srdci MTC onkogenní mutací RET, který se nachází na chromozomu 10q11.2. RET gen kóduje membránový receptor tyrosin kinázy, který ligandy patří do skupiny neurotrofních faktorů produkovaných gliových buněk (GDNF). Tento receptor je exprimován během vývoje v migrujících buněk neurální lišty, které dávají neuroendokrinní buňkám náběhem (C-buněk a buněk dřeně nadledvin) a buňky parasympatiku a ganglií sympatiku a periferního nervového systému. Různé mutace v RET jsou zodpovědné za pět různých syndromů. V srdci PA MEN MTC a rodiny jsou některé aktivační mutace v tomto genu. Ostatní aktivační mutace vést k MAN PB. Více než polovina z případů sporadické MTC dochází klonální somatické mutace (m. E. detekovatelný pouze v nádorových buňkách), totožná s jednou mutací způsobujících Familiární formy MTC. Genové přeskupování RETC vzniku chimérického genu je často pozorována v buňkách papilární rakoviny štítné žlázy. Experimenty s transgenních myší ukazují, že restrukturalizace genu RET je dostatečná pro vývoj rakoviny papilární štítné žlázy. A konečně, inaktivující mutace v RET způsobit, Hirschsprungova choroba - vrozené absence parasympatické ganglia břicha doprovázené porušení střevní motility, což vede k gigantismus tlustého střeva (megakolon).
Video: Nová éra v léčbě pokročilého medulární rakoviny štítné žlázy
Klinické projevy různých endokrinních syndromů neloplazii
MEN I
Hyperplazie příštítných tělísek (velmi často)
Slinivky (benigní nebo maligní)
- Gistrinoma
- inzulinom
- Glukagonomy, VlPomie (oba vzácné)
tumorů hypofýzy
- Sekretující růstový hormon
- prolaktin sekretující
- ACTH vylučující
Jiné nádory: lipom, karcinoid, nadledvinky a štítná žláza adenom
MEN IIA
Medulární karcinom štítné žlázy
Feochromocytom (benigní nebo maligní)
Hyperplazie příštítných tělísek
MEN IIB
Medulární karcinom štítné žlázy
feochromocytom
Neuromas sval ganglioneuroma
Marfanopodobnaya vzhled
Hyperparatyreóza (velmi zřídka)
} {Modul direkt4
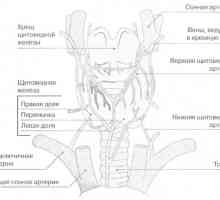
MTC je obvykle lokalizován ve středu nebo v horní části laloků štítné žlázy. V ojedinělých případech se obvykle objevuje v pouze v jednom ze segmentů, ale často multicentrických a bilaterální Kdy rodinné formy. Histologicky MTC dříve odlišuje od jiných typů rakoviny štítné žlázy přítomností amyloidních (eosinofilní látky, barvené Kongo červeně). Pak se ukázalo, že amyloid se skládá z husté fibrilárních proteinových depozit, které tvoří složenou vrstvu (-structure). Když takový protein je MTC prokalcitoninu nebo kalcitonin sám. Z tohoto důvodu, morfologická diagnóza MTC stanovena imunohistochemickým barvením na kalcitonin.
MTC se vyskytuje v různých způsobech. Sporadické nádory jsou oba velmi agresivní a extrémně maloaktivnymi- 5-leté přežití je 50%. Co se týče rodinných forem, to záleží na nich syndromy. Nejagresivnější MTC se vyskytuje u syndromu MEN IIB- přežít ne více než 50% pacientů během 2 let. Na MEN IIA pro MTC podobá sporadických forem rakoviny. Nejméně agresivní toky izolované familiární MTC. Nádor může šířit do regionálních lymfatických uzlin nebo dát hematogenní metastázy do plic a dalších orgánů. Metastazující nádor někdy doprovázené chronickým průjmem, patogeneze, která je nejasná. Kromě kalcitonin, tyto nádory vylučují řadu dalších biologicky aktivních látek, včetně prostaglandinů, serotonin, histamin a peptidové hormony (ACTH, somatostatin, kortikotropin uvolňující hormon). V některých případech je průjem odstraňuje dlouhodobě působící analog somatostatinu (oktreotid), který blokuje vylučování těchto látek.
Kalcitonin - MTC značky. Větší spolehlivost markeru dostane stimulaci sekrece. Typicky je to provedeno pomocí pentagastrinem (0,5 ug / kg iv 5 sekund) nebo rychlou infuzí glukonátu vápenatého (2 mg / kg po dobu 1 minuty). Vzorky krve byly odebrány před a po 1, 2 a 5 minut po stimulaci. Pro zvýšení citlivosti testu provádí kombinované: vápníku po infuzi okamžitě podávána pentagastrinu. V raných fázích nádorového bazální hladiny kalcitoninu je často normální, ale v metastatického MTC počáteční koncentraci kalcitoninu je několikanásobně vyšší, než je obvyklé. I přes to, že obsah vápníku v séru se nemění. Nádor se obvykle vylučuje kalcitonin forma vyšší molekulovou hmotnost a nižší biologickou aktivitu, ale často zvyšuje hladinu monomeru a kalcitonin. Ve většině případů, místo analýzy stimul vzorku provádí onkogenem RET, ale bazální kalcitonin udržuje svou hodnotu jako indikátor aktivity tumoru.
Všichni členové rodiny pacienta na podporu RET mutací by měli být vyšetřeni na MTC a jeho spojení s syndromů MEN IIA a IIB MEN. Při identifikaci mutace tohoto genu obvykle doporučují celkový tireoidekto-mise před vznikem rakoviny nebo zvýšit hladinu kalcitoninu. Děti v rodinách se známou mutací v RET je doporučeno, aby okamžitě vyšetřovat mutace nosič po narození. Po detekci mutací se provádí celkový tyroidektomii s lymfadenektomie, a v nepřítomnosti mutace je nutný další výzkum. Nicméně, je časování profylaktické štítné žlázy v případech asymptomatických nosičů mutací RET závislá na přesném genotypu (m. E. Mutantní kodon genu) a klinických podmínek. V případě vysoce rizikových specialistů doporučujeme provést operaci, dokud 6-12 měsíců věku, s průměrným rizikem - až na 5 let a při nízké štítné žlázy rizik může být odloženo až na 10 let. Jako základ pro více než 95% případů dědičné MTC a 25% sporadických nádorů je omezený počet genových mutací v RET, může být studie provedeny za použití standardních výstroje.
Studie rodinným příslušníkům musí být i se zřejmým sporadickým MTC, neboť téměř 25% nových případů probanda patří do rodiny s přepravou dědičného syndromu. Stejně jako ve známém rodinném MTC v blízké příbuzné provokačních zkoušek na sekreci kalcitoninu nebo analyzoval DNA v nádorových buněk a jiných buněk pro mutací RET. Identifikace mutací v nádorové tkáni naznačuje pouze jeho somatických a nádor - sporadické. Přítomnost stejné mutace v nádoru, a genomové DNA označuje familiární formu onemocnění a vyžaduje pečlivé zkoumání všech členů rodiny.
 Propojení malformace a nádory. Dědičné příčiny dětských nádorů
Propojení malformace a nádory. Dědičné příčiny dětských nádorů Hepatocelulární karcinom a rakovina tlustého střeva u dětí. Nádory štítné žlázy a nadledvinek
Hepatocelulární karcinom a rakovina tlustého střeva u dětí. Nádory štítné žlázy a nadledvinek Klasifikace rakoviny štítné žlázy. Papilární (papilární) adenokarcinom
Klasifikace rakoviny štítné žlázy. Papilární (papilární) adenokarcinom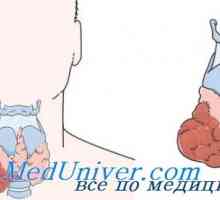 Dlaždicového (neorogovevayuschy) karcinom štítné žlázy. Nizkodifferentsirovaniaya forma rakoviny…
Dlaždicového (neorogovevayuschy) karcinom štítné žlázy. Nizkodifferentsirovaniaya forma rakoviny…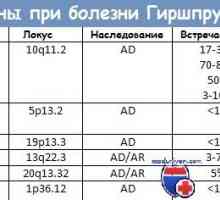 Patogeneze Hirschsprungova choroba embryogeneze, morfogeneze
Patogeneze Hirschsprungova choroba embryogeneze, morfogeneze Porušování migrační střevního nervového systému v Hirschsprung nemoci
Porušování migrační střevního nervového systému v Hirschsprung nemoci Příznaky rakovina štítné žlázy a příčiny, léčba a prevence rakoviny štítné žlázy
Příznaky rakovina štítné žlázy a příčiny, léčba a prevence rakoviny štítné žlázy- Vědci zjistili gen spojující rakoviny prsu a štítné žlázy
- V Evropě schválila nový lék pro léčbu rakoviny štítné žlázy lenvima
- Vliv mutace v ret efektivnosti terapie medulární karcinom štítné žlázy
 Štítné žlázy a příštítných tělísek
Štítné žlázy a příštítných tělísek- Štítné žlázy a příštítných tělísek. vrozená struma
- Syndrom vícenásobné endokrinní neoplazie typu 1
 Diagnostika a léčba rakoviny štítné žlázy
Diagnostika a léčba rakoviny štítné žlázy- Nádory. Endokrinní nádorová onemocnění přírodě cm. Akromegalie, virilnoe syndrom,…
- Je to o 1% všech nádorů. U žen se vyskytuje ve 2 krát častěji než muži. Průměrný věk nástupu 43-44…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
 Autoimunitní onemocnění štítné žlázy
Autoimunitní onemocnění štítné žlázy Syndromem mnohočetné endokrinní neoplazie, typ II (Maine 2): příčiny, příznaky, léčba, symptomy
Syndromem mnohočetné endokrinní neoplazie, typ II (Maine 2): příčiny, příznaky, léčba, symptomy Nádory neuroendokrinních buněk (koncept systému apud)
Nádory neuroendokrinních buněk (koncept systému apud)