Porušení metabolismu aminokyselin

Fenylketonurie (F) je způsobena deficitem enzymu fenylalaninhydroxylázy který konvertuje fenylalanin na tyrosin.
Dědičnost je autozomálně recesivní. Další varianty F (hyperfenylalaninemie IV a V), z důvodu nedostatku kofaktoru v této reakci. Onemocnění je doprovázeno vývojem hluboké mentální retardace, záchvatů, vyrážka, vysoké hladiny fenylalaninu v krvi a jeho metabolit fenylpyrohroznové kyseliny v moči, vůně myší moči. Prevence zahrnuje postnatální vyšetření a speciální dietu. Dědičná tyrosinémie (NT) je způsobena deficitem cytosolové fumaryl-acetacetátového hydrolázy, která katalyzuje první krok degradace tyrosinu. Mutantní gen mapuje na 15q23-Q25. Dědičnost je autozomálně recesivní. Dědičné tirozemiya typu I se vyskytuje u 1 dítěte 000-200 na 100 000 živě narozených dětí. Akutní tyrozinemie se vyskytuje v prvních týdnech života a je doprovázena vyčerpání, zvracení, průjem, hepatomegalie.
V chronických tvoří hlavní cílové orgány - játra a ledviny. Játra se zvětší, restrukturalizaci psevdozhelezistoy, progresivní fibróza, cirhóza transformace do zvýšené riziko vzniku hepatocelulárního karcinomu. Ledviny se může zvýšit, došlo k rozšíření kortikálních kanálků s jejich fokální kalcifikace. Možná kalcifikace cévní stěny, hyperplazie Langerhansových ostrůvků, hypofosfatemická křivice. V kožní biopsie na typ HT II označené akantózou nebo hyperkeratóza, elektronová mikroskopie - lipidů vakuoly průměru 2-3 nm a 10 nm mikrovláken ve spojení s údaji myelin. Lethal výsledek dochází v důsledku encefalopatie, meningitida nebo selhání jater.
Homogentisuria dědí autosomálně recesivní. Zpráva ve 3. kroku vede k hromadění vedlejších produktů metabolismu tyrosinu a fenylalaninu, kyseliny homogentisové, který propůjčuje tmavou barvu moči (homogentisuria). Kyslíkaté produkty této kyseliny, uloženého pod melaninopodobnyh pigmentů v chrupavky a vazivové tkáně, což představuje modro-šedé kůže, skléry a černé zbarvení klouby, s rozvojem artritidy v dospělosti (ochronosis). Pigment může být uložen v endokardu a srdečních chlopní hrbolky. Časné příznaky nemoci - pleny skvrna černé. V případě porušení poslední fáze metabolismu tyrosinu ke zvýšení hladin tyrozinu a methioninu v krvi, což může způsobit vývoj akutního selhání jater nebo vedou k rozvoji Fanconiho syndrom s poruchou funkce ledvin.
Video: MVI_0270.MOV
Tyrosin je zdrojem syntézy melaninu pokožky, vlasů, oka membrány. Je známo, po dobu nejméně 10 okulokutannogo albinismus syndromů s poruchou obsahu melaninu. Kromě úplné nebo částečné albinismus u pacientů s výraznou světelnou přecitlivělostí, snižuje ostrost vidění, oční svaly dysfunkce. Dědičnost je autozomálně dominantní, recesivní nebo sex-spojený. Kromě toho, jako prekurzor aminokyseliny hormonů štítné žlázy, blokáda metabolismu mohou být doprovázeny rodiny hypotyreóza, strumy, zakrnělý růst a duševní rozvoj.
Homocystinurie dědí autosomálně recesivní. Akumulace homocysteinu, cystein a dalších metabolitů je spojena s jedním z následujících mechanismů: nedostatek cystathionin (3-syntázy, methylcobalamin metabolické poruchy, poruchy methionin.
Video: Laminin - produkt aminokyselina
Typicky, pacienti s vysokým růstem funkcí marfanoidnymi, dislokace čočky, osteoporóza, mentální retardace, náchylný k tromboembolie velkých a malých krevních cév a rozvoje mozkových infarktů a vnitřních orgánů. Stěny tepen je označen fibrózu a ztluštění intimy, rozdělovat média svalových buněk a fragmentace elastických vláken, která předurčuje k předčasné těžkou aterosklerózy.
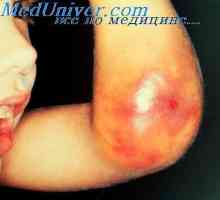 Afibrinotenemiya angiohemophilia a novorozenci. Hemoragické onemocnění novorozenců
Afibrinotenemiya angiohemophilia a novorozenci. Hemoragické onemocnění novorozenců Prekancerózních syndrom děti. dědičná onkogenezi
Prekancerózních syndrom děti. dědičná onkogenezi Akromezomelicheskaya dysplazie. eykardi syndrom
Akromezomelicheskaya dysplazie. eykardi syndrom Dominantní a recesivní alely chromozomů. dědičnost Autozomální dominanty
Dominantní a recesivní alely chromozomů. dědičnost Autozomální dominanty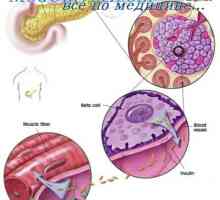 Autozomálně recesivní dědičnost. dědičnost X-vázaná
Autozomálně recesivní dědičnost. dědičnost X-vázaná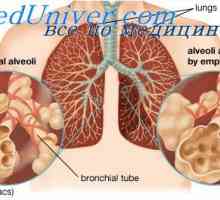 Uolmana zobecnit xanthelasmatosis onemocnění. refzuma syndrom
Uolmana zobecnit xanthelasmatosis onemocnění. refzuma syndrom Albinismus. Abderhaldenově cystinóza nemoc - ligmak - Kaufman a homocystinurie
Albinismus. Abderhaldenově cystinóza nemoc - ligmak - Kaufman a homocystinurie Galaktosemie
Galaktosemie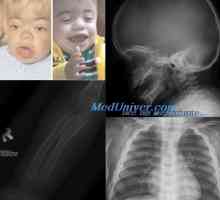 Klasifikace mukopolysacharidózy. Hurler nemoc, gentera, Sanfilippo, Morquio
Klasifikace mukopolysacharidózy. Hurler nemoc, gentera, Sanfilippo, Morquio Glykogen spalničky skladování choroba, Andersen McArdl. Hersey nemoc, Thomson, kontejnery
Glykogen spalničky skladování choroba, Andersen McArdl. Hersey nemoc, Thomson, kontejnery Autosomálně recesivní polycystické onemocnění ledvin u dětí. Diagnostika a léčba
Autosomálně recesivní polycystické onemocnění ledvin u dětí. Diagnostika a léčba Žloutenka u dítěte s tyrozinemie
Žloutenka u dítěte s tyrozinemie Renální tubulární acidóza. Proksimalnokanaltsevy
Renální tubulární acidóza. Proksimalnokanaltsevy Ataxie teleangiektaticheskaya (Louis-Bar syndrom), je relativně vzácné onemocnění s autozomálně…
Ataxie teleangiektaticheskaya (Louis-Bar syndrom), je relativně vzácné onemocnění s autozomálně…- Fenylketonurie závažné dědičné onemocnění, které se vyznačuje především zapojení nervového systému.…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
 Metabolické poruchy cyklu kyseliny močové
Metabolické poruchy cyklu kyseliny močové Vrozené makulární změny
Vrozené makulární změny Monogenní syndromy mvpr
Monogenní syndromy mvpr Metabolismus sacharidů u kojenců
Metabolismus sacharidů u kojenců Organické acidurie
Organické acidurie