Vrozené vady průdušky

Atrézie průdušky.
Vrozené atrézie průdušek je vzácný, ale u novorozenců a je obvykle v kombinaci s infantilní lobární emfyzém. V 90% případů v levém horním rohu, pravém horním rohu a pravého středního laloku. Nejvíce postiženy segmentového průdušek, ale může být ovlivněna subsegmentary a spravedlnost. Příčinou průdušek atrézie u novorozenců je nitroděložní zánět. Někteří autoři poukazují na to, cévních chorob v genezi zla. Mikroskopicky: ve tvaru prstence nebo excentrický fibróza bronchiální stěny, s abnormalitami chrupavčitých destiček, nebo bez nich. Někdy je pás vazivové tkáně v místě atrézie nebo lumen naplněné relativně normální sliznicí. Distální část průdušky atrezirovannogo cystické rozšířené, naplněné hlenu a připomíná mukokély.
bronchostenosis. Příčiny kongenitální stenóza jiný - fibrózou stěny ve výsledku nitroděložního zánětu, ektopie tkáň štítné žlázy průduškového stěny, vrozené sliznice, stejně jako stlačení průdušky mimo parabronhialnymi hmot, např teratom nebo bronchogenního cyst, zvýšené nebo abnormálně rozložených plicní tepny, zvýšená levé nebo pravé předsíně , průdušek stenóza může být spojena s infantilní lobární emfyzém, jícnu atrézie a tracheoesofageální píštěle.
Abnormální větvení a vykašlávání z průdušek. Existují různá provedení anomální vypouštění větvení a lobární průdušky, v 75% případů - průdušnice (trachey průduškového). Může skončit naslepo. V odpovídajícím plic zóně se vyvíjí zápal plic.
Bronhobiliarny fistule. To představuje vzácnou vadu, ve kterém pravý hlavní průdušky a levý jaterní potrubí připojené píštěle, která se rozprostírá podél jícnu přes membrány. Embryologický Základem tohoto zla není zcela jasné, ale může to být formou duplikace v horní části gastrointestinálního traktu. Proximální část píštěle ve struktuře se podobá bronchitidu s respirační epitel a chrupavčitých prstenců a distální - žlučovodu. Klinicky pozorována opakující se zánět plic, sputum barevný žluč.
Bronhomalyatsiya. Nejčastěji vidět u předčasně narozených dětí, kteří byli po dlouhou dobu na ventilátoru. Nicméně, zde je vlastní a bronhomalyatsiya způsobil bludný vývoj chrupavky průdušek, což vede ke zhroucení dutiny a vývoj sekundární pneumonie. Tato vada je popsána jako součást syndromu recesivní rodiny X-vázanou charakterizované deformaci hrudní stěny, mentální retardace, dolichocephaly, svalová dystrofie kryptorchizmu a testikulární atrofie. Mikroskopicky: postižená průduškový je snížena velikost, mezi normální chrupavčité desky jsou umístěny ostrůvky nezralá chrupavka. V plicní vzdáleném od postiženého průdušek - záněty nebo infantilní lobární emfyzém. Změny v průduškách mohou být kombinovány s podobnou patologii chrupavky průdušnice (traheobronhomalyatsiya).
vrozená bronchiektázie. Popsáno v mnoha vrozených i rodinným poruch, včetně syndromů deficitu imunity (nedostatek neutrofilů a komplementu) a selhání, antitrypsinu, primární ciliární dyskinesie, cystická fibróza, Kartagener syndrom, Svayera - James (jednostranné supertransparency plic) a Williams - Campbell.
Williams syndrom - Campbell - autosomálně recesivní dědičná forma provedení onemocnění bronhomalyatsii vyznačující se chrupavka hypoplazie segmentové kroužky a subsegmentární průdušky. Tento syndrom se klinicky projevuje u novorozenců a kojenců. Děti s rozsáhlými bronchiektaziemi umírá krátce po narození asfyxie jevů. Omezené formy zlozvyku v novorozeneckém období ve většině případů zůstává nezjištěno.
Video: Vrozené vady
Primární ciliární dyskinesie a Kartagener syndrom (starý název - pevné řasinky syndrom) -autosomno recesivní dědičné onemocnění způsobené dëdicnych ultrastruktury řasinkami epitelu. Nejčastější a klasický tvar primární ciliární dyskineze Kartagener syndrom je charakterizován triádou příznaků: nepřímé uspořádání vnitřních orgánů (Situs viscerus inversus), bronchiektázie a zánět vedlejších nosních dutin. Předpoklad, byla provedena o vztahu mezi polohou vnitřních orgánů a stanovení funkce ciliární v děloze. Bez ciliární funkce umístění těl náhodné (v 50% případů normální uspořádání vnitřních orgánů a v 50% - inverze). Důsledkem ciliární dyskinesie stagnuje sekrety respiračního traktu, infekce, tvorba bronchiektázie a chronického zánětu (zánět vedlejších nosních dutin, zánět průdušek, zánětu středního ucha, jsou tvořeny 15% pacientů s nosní polypy).
Řasinky vady diagnostikované elektronovou mikroskopií. Mezi ně patří: nepřítomnost nebo zaostalosti vnitřních a vnějších rukojeti dineinovyh, nedostatek radiálních paprsků, nepřítomnost jednoho nebo obou centrálních mikrotubulů.
Videa: 10 nejstrašnější vrozených vad
U většiny pacientů porušením respiračních funkcí pozorované v novorozeneckém období, ale bronchiektázie a nosních polypů zjištěny později, když v prvních letech života, i ve starším věku. V posledních letech, dineinovye poruchy spojené s mutací v oblasti krátkého ramene chromozomu 9 (9p21-P13).
Bronchiální (plicní), syndromy jsou isomerní. Plicní izomerie - patologie, ve kterém abnormální rozvětvení průdušek vede k tvorbě z obou stran dvou totožných lehkých, tj. dvudolevogo "vlevo" a tři-share "right". Ve většině případů, to je v kombinaci s MVPR nejčastěji - s srdečních vad a sleziny. Výskyt isomerních syndromů kvůli neschopnosti raného embrya se vytvořil pravolevou asymetrii. V literatuře mnoho SVYHLEDAT syndromy izome-ismus, nejznámější pět typů syndromů s izomerovaných-IOM a sleziny poruch.
Video: Kategorie "PRO zdraví": Vrozené vady
- Typ I - syndrom, asplenií Ivemarka - Jednotlivé komplexní vrozené vady s pravým izomerie, včetně nedostatku sleziny, špatným směrem střeva, střední polohy jater, bilaterální trehdolevym „správnou“ světla se dvěma hlavními průdušek, která se nachází nad plicní tepny (obvykle vpravo průdušky je přes plicnice, levá - pod plicní tepně) a závažné srdeční vada: VSD a ASD, mezo a dextrokardie, atrioventrikulární komunikace, transpozice velkých cév, atrézie nebo stenóza plicní ostrý th barel a kol., s výhodou ovlivňuje chlapců.
- Typ II - M-anizospleniya - se vyskytuje pouze u mužů (tedy zkratku - M - v tomto typu vady titulu). V tomto případě, jsou: relativně normální zobrazení vnitřních orgánů, bilaterální dvudolevoe „vlevo“ světlo se dvěma lobární průdušky, který se nachází nad plicní tepny, vrozené vady srdce a anizospleniya (přítomnosti jednoho nebo více větších slezin nebo jeden nebo více menších sleziny).
- Typ III - polysplenia syndrom - vyznačující se tím, nesprávné umístění vnitřních orgánů s poruchou střevní otáčení, mediální umístění jater, vrozené vady srdce (defektu síňového septa a měničem, bilaterální morfologicky levé síně) a polysplenia: přítomnost násobku (4-14) jsou rovnoměrně malé sleziny. K dispozici je levostranná izomerie s oboustranným dvudolevym snadné s obou hlavních průdušek, která se nachází pod plicnice.
- Typ IV-0-anizospleniya - označené abnormality méně výrazné viscerální orgány, než když asplenií a polysplenia. V centru - pravá komora dvojvýtoková. Anomálie plic a sleziny jsou stejné jako u typu 3 se týká pouze dívky.
- Type-0 V-anizospleniya - levostranná plic izomer. Srdce - pravá komora dvojvýtoková General atrioventrikulární ústí vada nebo obojí. V 50% případů - abnormální intestinální otáčení, více slezin. Chlapci a dívky jsou postiženy stejně často.
Ve většině z těchto syndromů jsou sporadické, ale je popsáno autozomálně recesivní, autosomální dominantní a X-vázané typy dědičnosti.
 Mechanismy hrtanu atrézii. Uzi hrtanu atrézie
Mechanismy hrtanu atrézii. Uzi hrtanu atrézie Prognóza a léčba hrtanu atrézie. Chylotorax u plodu
Prognóza a léčba hrtanu atrézie. Chylotorax u plodu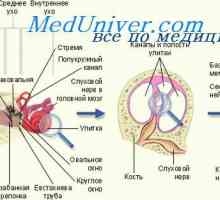 Průdušky embryo. Fetálního vývoje průdušky
Průdušky embryo. Fetálního vývoje průdušky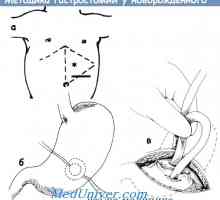 Indikace a kontraindikace k gastrostomií novorozence
Indikace a kontraindikace k gastrostomií novorozence Tísňová péče v traheoezofagealnoy píštěle u novorozenců
Tísňová péče v traheoezofagealnoy píštěle u novorozenců- Klinická anatomie průdušnice a průdušek
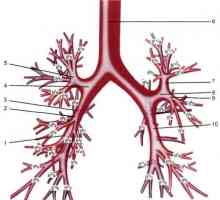 Anatomie velkých průdušek
Anatomie velkých průdušek Průdušnice a průdušek švy
Průdušnice a průdušek švy- Plicní žíly, vpravo a vlevo venae pulmonales dextrae et sinistrae, vyjmout arteriální krve legkih-…
- Nebo saccular bronchiektázie válcového výstupku segmontarnyh subsegmentární průdušek a chronické…
- Průdušek cizí těleso často dostat v pravém vzdálenějším průdušky. Historie aspiraci cizího tělesa…
- Atrézie anu mohou způsobit pozdní diagnóza při nízké ileus. anální atrézie je často spojován s…
- Chirurgická onemocnění hrudníku. jícnu atrézie, závažné malformace, který je vytvořen v časných…
- Průdušek cizí tělesa v 80% případů spadá do pravé průdušky být téměř přímým pokračováním…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Bronchiektázie (bronchiektázie)
 Anatomie hlavní průdušky. cévní zásobení a inervace hlavní průdušky
Anatomie hlavní průdušky. cévní zásobení a inervace hlavní průdušky Tuberkulóza nitrohrudních mízních uzlin: léčbu, komplikace, příčiny, příznaky
Tuberkulóza nitrohrudních mízních uzlin: léčbu, komplikace, příčiny, příznaky Jícnu atrezie novorozenců: efekty, příčiny, příznaky, léčba, symptomy
Jícnu atrezie novorozenců: efekty, příčiny, příznaky, léčba, symptomy Vrozené vady hrtanu a průdušnice
Vrozené vady hrtanu a průdušnice