Vrozená a dědičná tubulopatie u dětí
Video: Příčiny autismu: genetika, genová autismu, vakcína DTP, gluten

Tubulopatií - onemocnění s primárními lézemi přední trubkovitý, v němž rozbité membránový transport různých látek v ledvinových tubulů.
Video: instinkty, děti, stres, vzdělání - Jacque Fresco - Projekt Venus
U novorozenců zjištěných klinicky izolovaný tubulopatie, které jsou většinou doprovázeny polyurie a poruchy elektrolytů v plazmě. Patří mezi ně Bartterův syndrom, Liddle, Lowe, diabetes insipidus, primární typ gipoaldosteronizm I, hyperkalcemie „raného dětství“ nebo trubkového typu acidóza IV. S výjimkou Bartterova syndromu a Lowe, morfologické změny v ledvinách při chybějící nebo charakterizované rozvojem nefrokalcinózy těchto tubulopatie.
Video: diagnóza vrozené a dědičné patologie Seminář
Bartterův syndrom - tubulopatie, hypokalémie projevuje chloropenia, metabolická alkalóza a hyper reninemiey s normálním krevním tlakem. Existují tři formy syndromu: neonatální, klasické a Gitelmanova syndrom (u novorozenců nebyl nalezen). Novorozenecké a klasické Bartterovým syndromy někdy zdědil autozomálně recesivní, ale ve většině případů sporadické. Které lze identifikovat podle dvou genotyp podtypu novorozeneckých formy syndromu. Podtyp I způsobena mutací v genu kotransportnom sodný a chlorid draselný (SLC12A1 lokusu na dlouhém rameni chromozomu 15-15q15-21), subtypu II - ROMK mutace v genu (KCNJ1 lokusu na dlouhém rameni chromozomu 11 (11q24-25) pro klasický Bartterova syndromu. vyznačující se tím, mutace v genu pro sodíkových kanálů (lokus CLCNKB na krátkém raménku chromozomu 1-1r36). v obou formách tam Polyhydramnios matkám a dětem, často se rodí předčasně, při narození může být masivní polyurie. při studiu plodové vody v prenatálním období je detekován SW lichenie obsah chloridu, jak bylo určeno ultrasonografie nefrokalcinózy, hydronefrózy a někdy v důsledku chronických hydroureter polyurie.
Novorozenecká forma je diagnostikována brzy po narození, klasické - u novorozenců a kojenců ve věku do dvou let.
Video: Vrozená onemocnění dětí - je výzvou pro rodiče?
Mikroskopicky pozorováno, spolu s hyperplastické nefrokalcinózu juxtaglomerulárního aparátu (patognomonické značka), méně často - hyperplazie, medulární intersticiálních buněk, někdy - hyalinóza glomerulární vakuolizace apikálních epitelových kanálků, tubulární atrofie a intersticiální fibrózy. Úmrtnost a mortalita v neošetřené Bartterova syndromu. Při léčbě stavu dítěte výrazně zlepšil, ale dlouhodobá prognóza zůstává nejistý, protože postupně postupuje pomalu CRF.
Lowe syndrom (syndrom oculocerebrorenal) - vzácná genetická choroba je X-vázaná recesivní způsob dědičnosti. Syndrom je způsobena mutací genu OCRL1 umístěn na chromozomu X (Xq25-26). Ženy - nositeli abnormálního genu šedý zákal, ale mají normální renální a neurologické funkce. Toto onemocnění se vyznačuje kombinací vrozené katarakty a glaukomu, těžké mentální retardace, hypotonie s renální tubulární dysfunkcí.
Intenzita a doba tubulární dysfunkce jsou variabilní. Renální funkce a struktura při narození může být normální. Proximal trubička dysfunkce často začíná ve věku 3-12 měsíců, mezi tím. Mikroskopická struktura ledvin u plodů a novorozenců je obvykle normální. Avšak již v prvních měsících života jsou pozorovány expanzi tubulů, které atrofii a proteinové válce v lumen. Glomerulů od normálních novorozenců a pak se vytváří sklerózu (fokální nebo globální glomeruloskleróza), GBM zahuštěný. Elektronová mikroskopie uvedené fúze nohy podocytů a tubulární poškození mitochondrií, ale není jasné, zda se jedná o primární ve vztahu k trubkové dysfunkce.
 Galaktosemie
Galaktosemie Resuscitační v hypofosfatémie
Resuscitační v hypofosfatémie- Resuscitační v hypokalémii
- Autismus naučit identifikaci dětí šestiměsíční
 Diabetes insipidus: příčiny, klinický
Diabetes insipidus: příčiny, klinický Distalnokanaltsevy acidózy u dětí. klinika
Distalnokanaltsevy acidózy u dětí. klinika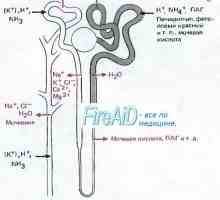 Renální tubulární acidóza. Proksimalnokanaltsevy
Renální tubulární acidóza. Proksimalnokanaltsevy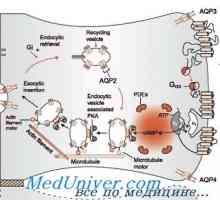 Křivice s tubulární acidózy. Diabetes insipidus u dětí
Křivice s tubulární acidózy. Diabetes insipidus u dětí- Gitelmanova syndrom u dětí. Diagnostika a léčba
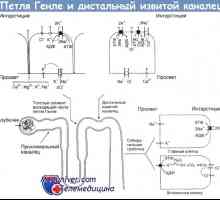 Vyměnit syndromu u dětí. Diagnostika a léčba
Vyměnit syndromu u dětí. Diagnostika a léčba Chronická tubulointersticiální nefritida u dětí. Diagnostika a léčba
Chronická tubulointersticiální nefritida u dětí. Diagnostika a léčba- Diabetes insipidus
- Hyperlipemie essontsialnaya (gepatosplenomega krystaly lipoidoz) -skupinu dědičné fermentopathia…
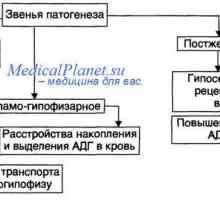
- Diabetes insipidus, onemocnění způsobené absolutním nebo relativním nedostatkem antidiuretického…
- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
- Léčba onemocnění ledvin,
 Pseudohyperaldosteronismus
Pseudohyperaldosteronismus Renální tubulární acidóza: symptomy, příčiny, léčba, symptomy
Renální tubulární acidóza: symptomy, příčiny, léčba, symptomy Fanconiho syndrom: Příznaky, léčba
Fanconiho syndrom: Příznaky, léčba Jiné patologie novorozenců
Jiné patologie novorozenců Příštítných tělísek u dětí
Příštítných tělísek u dětí