Dědičných chorob, které jsou typické pro určité etnické skupiny

Dědičných chorob, které jsou typické pro určité etnické skupiny.
Již jsme uvedli, že v určitých etnických skupin, se vyskytují některé genetické choroby častěji:
- v Afro - srpkovitá anémie a thalassemia;
- u osob ze Středomoří - talasemie;
- ve frankofonní Kanaďané - Tay-Sachs nemoc;
- Respondenti z Velké Británie a Irska, z Jižní Ameriky a ze severní Číně - defekty neurální trubice;
- White - cystická fibróza,
- V Ashkenazi Židé - Tay-Sachs nemoc, Canavan, cystická fibróza a Gaucher.
Až na defekty neurální trubice (typicky polygenní onemocnění), které se dědí autozomálně recesivní. U každého z nich vyvinula diagnostické metody.
Pro identifikaci srpkovitou anémií, jakož i - a thalassemia provést kompletní krevní obraz a elektroforéza hemoglobinu. V srpkovitá anémie, hemoglobinu pomocí elektroforézy může odhalit globinu genového produktu, a-řetězec s bodovou mutací, jakož i další klinicky relevantních řetězci globin variant, včetně hemoglobinu S. normálního dospělého hemoglobin obsahuje hemoglobinu A1 (přibližně 98%) a hemoglobinu A2 (2-3%). Thalassemia vede k mikrocytární anemie, avšak střední korpuskulární objem menší než 80 mikrometrů3 může naznačovat vozík - nebo -talas-SEMII. Při zvýšené hladiny hemoglobinu A2 (3,5%) by se předpokládat, talasemie. Za normálních výsledků elektroforézy hemoglobinu v kombinaci s mikrocytózy, které nejsou způsobeny nedostatkem železa by mělo být podezření talasemie. Pro potvrzení diagnózy ukazují na přítomnost genu delece a-globin řetězec molekulárně-genetických metod.
Pro diagnostiku dalších onemocnění vyskytující se v určitých etnických skupin používají biochemické nebo molekulárně genetických metod. Tay-Sachs nemoc obvykle detekovány podle úrovně hexosaminidázy sérum (těhotenství nebo příjem OK vyžaduje leukocytů kultury). V nejasných případech je také provedena analýza DNA. V Ashkenazi Židů se může provádět přímo na průzkumu řada charakteristických dědičných chorob: Tay-Sachsova choroba, Canavan, Gaucherovy a cystická fibróza - to se obvykle provádí pomocí molekulárně genetickými metodami.
Screening cystické fibrózy
Cystická fibróza - onemocnění s autozomálně recesivní způsob dědičnosti v bílém: se vyskytuje u novorozenců s frekvencí 1: 2500-1: 3300. Mutace v genu CF, který se nachází v lokusu 7q31 a kóduje protein, - membránové regulátor vodivosti, způsobují zvýšení viskozity plicních tajemství trávicí soustavy, reprodukčních orgánů, a potní žlázy. Smrt u cystické fibrózy dochází nejčastěji z poškození plic, a průměrná délka života jen zřídka přesahuje 30 let. Přibližně 85% pacientů také poznamenat, nedostatečnou exokrinní pankreatické funkce zhelezy- mezi další příznaky - chronické sinusitidy, nosní polypy, pankreatitida, onemocnění jater.
V poslední době se snažit zajistit, aby screening pro cystickou fibrózu se stává stále rozšířenější. Dříve to bylo nabídnuto v přítomnosti nemoci v nejbližší rodiny nebo svého partnera. Nicméně, jak rada se domnívá, že průzkum by měl být pro všechny těhotné ženy a všechny ženy, které plánují těhotenství, k dispozici - to bude určení možných nositelů mutovaného genu. V praxi to znamená, že screening cystické fibrózy by měla být nabídnuta všem se líbil lékařem o neplodnosti. Inspekci partneři mohou být postupně (první, v případě pozitivního výsledku - druhý) nebo současně. Pokud jsou nositeli mutantní alely budou mít oba partneři riziko narození pacienta s každým těhotenstvím je 25%.
Pozornost by měla být věnována lidem s bilaterální aplazií chámovodu: měly by být podezřelá cystickou fibrózu, dokud dokud se neprokáže obratnoe- proto je nutné zkoumat a partnerem. Zavedení hromadné screening cystické fibrózy by měla tento problém odstranit, protože budou muset kontrolovat všechny bez výjimky. Nicméně, některé páry se mohou odhlásit z průzkumu, a je-li člověk v tomto páru má bilaterální Aplazie chámovodu, musíte být trvalé, nebo alespoň mluvit o cystické fibróze těch, kdo trvá na svém odmítnutí. Dokonce i když člověk s aplazií chámovodu není zasažen světlo, musel pacientů s cystickou fibrózou, děti se mohou narodit.
CF gen má délku 230.000 párů bází a skládá se z 27 ekzonov- i když odhaleno více než 1000 různých mutací, ale doporučuje se pro kontrolu na přítomnost 25 nejběžnější (které se nacházejí v USA s frekvencí alespoň 0,1%), typické pro všechny etnické skupiny. White dominuje vypouští DO508 (60-70% všech případů). Mutace jsou zahrnuty v programu průzkumu, bez ohledu na to, zda světla nebo těžkou formou onemocnění, které způsobují. Mutace zjištěné pomocí PCR za použití alelově specifických oligonukleotidy- ve většině laboratořích automatizovaného postupu. Je třeba zdůraznit, že další analýzu na přítomnost dalších mutací (rozšířené inspekce program) četnost detekce cystické fibrózy není v současné době zvyšuje, a proto se nedoporučuje. Vzhledem k tomu, že mutace obsažené v seznamu jsou uvedeny ve Spojených státech s frekvencí, která není nižší než 0,1% veškerých dalších mutací bude vzácnější.
Některé alely genu CF (není příčinou cystické fibrózy), v některých případech může vést k falešně pozitivním výsledkům. Po detekci některých mutací (AF508 nebo D1507) v homozygotním stavu v zamýšleném nosiči (které nemůže být homozygotní) by měly být analyzovány na přítomnost alel F508C (způsobí, cystická fibróza) a I506V a I507V (nezpůsobuje onemocnění). Po detekci R177H mutace by měly být testovány na přítomnost alely 5T / 7T / 9T (viz. Níže, v části o dvoustranné aplazie vas deferens). Toto takzvané deriváty testy - jsou prováděny pouze tehdy, když určité mutace a nejsou součástí původního šetření. Analýza se musí provádět akreditované laboratoře provádějící diagnostické všech 25 mutací ze schváleného seznamu. Ve své zprávě, by laboratoř měla určit etnický původ subjektu, důvod k postoupení k analýze, seznam testovaného a mutační analýzou. V případě, že výsledek studie je pozitivní, musí zpráva obsahovat doporučení týkající se průzkumu partnerovi, a pokud je záporná - posouzení zbytkového rizika (s přihlédnutím, že analýza se nepodařilo detekovat mutace v genu CF způsobují cystickou fibrózou). Nosné frekvence alel, které způsobují cystickou fibrózu, mezi bělochy evropského původu je 01:29 a mezi Američany Afričana - 1:65. Screening pro cystickou fibrózu - technicky nesmírně složité laboratorní metody, tedy American College of Medical Genetics, American College of patologů a zvláštní komise pro genové diagnostice National Institutes of US Department of Energy zdravotnictví a vyvinuly doporučení na kontrolu kvality.
Samostatné diagnostické problémy spojené s oboustranným aplazií semenných-eferentní potrubí, ale je důležité si uvědomit, že to je cílem identifikovat ani při prohlídce na cystickou fibrózou. Pokud máte jakékoliv pochybnosti vzniklé při studiu zprávy laboratorní lékař měl poslat dvojici do genetiky. Kromě toho, genetické poradenství by měly být zaslány do každé z následujících případech: v případě, že pár, ve kterém pouze jeden partner je nositelem mutace, se chce dostat více informatsiyu- pokud mezi nejbližší příbuzní mají pacienti mukovistsidozom--li uvedeno jinak zdravých mužů identifikovaných mutací což vede k bilaterálním aplazií chámovodu (kromě případů, kdy androlog nebo specialista na léčbu neplodnosti sama o sobě může vypořádat s výsledky analýzy a dát pacient potřebuje výpis neniya) - je-li oba partneři jsou nositeli mutantních alel - v tomto případě, je třeba vědět o možném narození svého nemocného dítěte, pravděpodobnost toho a na možnosti budoucí chování.
 Pyruvát kináza nedostatek plodu. hemoglobinopatie
Pyruvát kináza nedostatek plodu. hemoglobinopatie Haemoglobinopatologií s. Thalassemia u novorozenců
Haemoglobinopatologií s. Thalassemia u novorozenců Ganglinarnaya deska embryo. Primární váčky mozkové
Ganglinarnaya deska embryo. Primární váčky mozkové Porušování migrační střevního nervového systému v Hirschsprung nemoci
Porušování migrační střevního nervového systému v Hirschsprung nemoci Anémie je to možné dědičné příčiny anémie
Anémie je to možné dědičné příčiny anémie- Anémie příčiny a druhy chudokrevnosti
- Nízké hladiny hemoglobinu způsobuje anémii
- Hemoglobinopathiemi v těhotenství: příčiny, léčba, symptomy
- Screening během těhotenství, genetický screening novorozenců
- Jaké jsou genetické choroby
- Nemoci hematopoetického systému. thalassemia
 Indikace k invazivní metody vyšetřování plodu.
Indikace k invazivní metody vyšetřování plodu. Načasování prenatální diagnostiku genetických chorob.
Načasování prenatální diagnostiku genetických chorob.- Hematologie-variabilita fetálního hemoglobinu v normální a hemoglobinopathies
- Anémie
- Thalassemia
- Genetické choroby jsou jedny z nejčastějších onemocnění
- Srpkovitá anémie: výzva pro FDA
 Hemoglobin (hemoglobinopatie)
Hemoglobin (hemoglobinopatie) Thalassemia, léčba, příznaky, příčiny, příznaky
Thalassemia, léčba, příznaky, příčiny, příznaky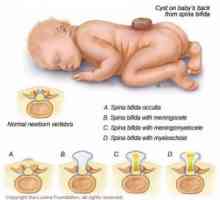 Neurální trubice vady u dětí: encephalocele, meningomyelocoele
Neurální trubice vady u dětí: encephalocele, meningomyelocoele