Gitelmanova syndrom u dětí. Diagnostika a léčba
Video: Diagnostika a léčení ADHD, d m n, profesorem .... LS Chutko
Gitelmanová syndrom (Často označovaných provedení, Bartterův syndrom), to je také vzácné autosomálně recesivní onemocnění s gipokaliemicheskoe metabolická alkalóza. Charakteristickým rysem syndromu - gipokaltsiuriya a hypomagnesémii, v typických případech se projevuje v pozdním dětství nebo v rané dospělosti.
Video: Léčba West syndromu. Klinika a diagnostika West syndromu
Patogeneze Gitelmanova syndromu. Biochemické změny v Gitelmanova syndromu se podobají těm při konstantní užívání thiazidových diuretik. Tyto prostředky působí na kotransportéru chloridu sodného, NCCT, lokalizovaných v distálních spletitých kanálků. Genetické studie ukazují, pacienti s defekty v genu kódujícího NCCT.
Klinické projevy Gitelmanova syndromu. Gitelmanova syndrom je obvykle projevuje později v životě, než Bartterovým syndromu. Pacienti mají často v minulosti svalové křeče a křeče důvodu, pravděpodobně nízké hladiny hořčíku v séru. Opakované epizody dehydratace jsou obvykle chybí. Biochemické změny zahrnují hypokalémii, hypomagnezémii a metabolické acidózy. Hladina vápníku v moči výrazně snížena (na rozdíl od Bartterova syndromu), a obsah hořčíku se zvyšuje.
Video: mozková obrna příznaky a léčba. Jak vyléčit Radislav. hydrocefalus

Úroveň reninu a aldosteron, obvykle norme- nezvyšuje sekrece prostaglandinu E. zakrnění je méně výrazné než u Bartterova syndromu. Diagnóza. Gitelmanova syndrom je diagnostikován u dospívajících nebo dospělých v přítomnosti gipokaliemicheskogo metabolickou alkalózu, hypomagnezémii a gipokaltsiurii. Léčbu. Pro korekci hypokalémie a použití hypomagnesémii draslíku a hořčíku doplňků. nejsou obvykle potřebné přísady sodný nebo inhibitory syntézy prostaglandinů, protože žádný hypovolémie, žádné zvýšení vylučování prostaglandinu E u pacientů, kteří nejsou obvykle pozorovány.
V současné době, vrozené vady vyznačující se liší transportéry, působí v různých částech nefronu. Renální tubulární acidóza a diabetes insipidus podrobně popsány v tomto pořadí. Autosomálně recesivní cystinurie je převážně pozorována u jedinců pocházejících ze Středního východu. Je charakterizována opakovanou tvorby močových kamenů způsobených vysokou afinitou transportér defektu L-cystin a dvojsytných aminokyselin v proximálních tubulech ledvin.
pro X-vázaná urolitiáza (Dent choroba) je také charakterizována opětovné tvorbě kamenů a progrese do Fanconiho syndrom. Podle jejího průzkumu X-vázanou chorobou se vyskytuje téměř souhrnně u chlapců. Je založena na mutaci genu kódujícího napětí citlivé kanály, CLN5, přítomný ve všech částech nefronu. Aktivaci nebo inaktivaci genu pro receptor mutace vápenatý (přes které PTH posiluje reabsorpci vápníku v Henleovy kličky) resp způsobit vážné hypo- a hyperparatyreózy.
aktivace mutace genu epiteliální sodíkový kanál pracující ve sběrných kanálků základem dědičné formy hypertenze, syndrom Lydd la. U pacientů s tímto syndromem konstitučně aktivovanou reabsorpci sodíku ve sběrných kanálků, snížené hladiny draslíku v séru a potlačuje sekreci aldosteronu. Inaktivující mutace v tomto genu základem Pseudohypoaldosteronism projevuje ostrý ztrátu sodíku a hyperkalemie. Varianta tohoto stavu se vyznačuje tím, systémových poruch, včetně vylučování potu chloridu, a může se podobat cystickou fibrózou.
 Dehet syndromu u plodu. Aase syndrom, Holt-Oram plod
Dehet syndromu u plodu. Aase syndrom, Holt-Oram plod Redukční defekty končetin plodu. Phocomelia plod
Redukční defekty končetin plodu. Phocomelia plod Fraser syndrom. Diagnostika a léčení syndromu u plodu Fraser
Fraser syndrom. Diagnostika a léčení syndromu u plodu Fraser Holt Oram syndrom. Gidroletalny syndrom
Holt Oram syndrom. Gidroletalny syndrom Frinsa syndrom. Diagnostiku a léčbu syndromu u plodu frinsa
Frinsa syndrom. Diagnostiku a léčbu syndromu u plodu frinsa Trenaunay Syndrome Klippel-Weber. Larsena syndromu u plodu
Trenaunay Syndrome Klippel-Weber. Larsena syndromu u plodu Syndrom smrtící pteriguma. Lissencephaly i typ
Syndrom smrtící pteriguma. Lissencephaly i typ Pfeiffer syndrom. Diagnostika a prognóza Pfeiffer syndromu
Pfeiffer syndrom. Diagnostika a prognóza Pfeiffer syndromu Morfologie, diagnostika a léčba syndromu Stein-Leventhal
Morfologie, diagnostika a léčba syndromu Stein-Leventhal Autosomálně recesivní polycystické onemocnění ledvin u dětí. Diagnostika a léčba
Autosomálně recesivní polycystické onemocnění ledvin u dětí. Diagnostika a léčba Autozomálně recesivní syndrom hyperproduction imunoglobulinu m (IgM). genová mutace pomoc
Autozomálně recesivní syndrom hyperproduction imunoglobulinu m (IgM). genová mutace pomoc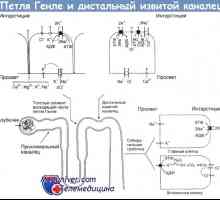 Vyměnit syndromu u dětí. Diagnostika a léčba
Vyměnit syndromu u dětí. Diagnostika a léčba- Peutz-Jeghersův syndrom způsobuje, příznaky a diagnostika Peutz-Jeghersova syndromu
- Alportův syndrom příčiny a příznaky, diagnostika a léčení Alportova syndromu
- Velokardiofatsialny syndrom
 Syndrom vlasy-an
Syndrom vlasy-an Syndrom dráždivého střeva, příznaky, léčba, příčiny, příznaky
Syndrom dráždivého střeva, příznaky, léčba, příčiny, příznaky Korsakova syndrom u alkoholismu: léčba, příznaky, prognóza
Korsakova syndrom u alkoholismu: léčba, příznaky, prognóza Noonan syndrom (syndrom muž Turner)
Noonan syndrom (syndrom muž Turner) Vrozená a dědičná tubulopatie u dětí
Vrozená a dědičná tubulopatie u dětí Bartterův syndrom a Gitelmanova syndrom u dětí: příznaky, léčba, příčiny
Bartterův syndrom a Gitelmanova syndrom u dětí: příznaky, léčba, příčiny