X-vázaná lymfoproliferativní syndrom chlapců. Duncanova nemoc
X-vázaná lymfoproliferativní syndrom, také volal Duncanova nemoc (Za rodiny, v níž byl poprvé identifikován) je X-vázaná recesivní znak vyznačuje sníženou imunitní odpovědí proti viru Epstein-Barrové.
Genetika a patogeneze X-vázanou lymfoproliferativní syndrom - onemocnění Duncan. Defektní gen je lokalizován na dlouhém rameni chromozomu X (stránky Xq23) a klonován. Zpočátku, jeho proteinový produkt byl jmenován SAP (SLAM-spojený protein), ale později dostal oficiální název SH2D1A. SLAM (signalizaci aktivace lymfocytů molekula - signalizace lymfocytovou aktivaci molekuly) je adhezní molekula.
Infekční činidla a další stimulanty urychlit jeho syntézy v T- a B-lymfocyty. SH2D1A je vysoce exprimován na thymocytů a T (nejlépe na Th1) a NK-buněk periferní krve. Není jasné, zda je přítomen v B-lymfocytů. Takže i když se onemocnění je často pozorováno selhání Duncan protilátek skutečně ovlivňuje základní vadu T- a NK-buněk. SH2D1A soutěží s SHP-2 o vazbu na protein SLAM, a tak působí jako regulační molekuly.
Nedostatek SH2D1A Určuje nekontrolované reakce cytotoxických T-lymfocytů virem infekce virem Epstein-Barrové. Z SH2D1A proteinu je závislá na expresi 2b4 molekul na NK-buněk. Porušení aktivace NK-buněk zprostředkovaných molekuly, také hraje roli při poruchách imunity během nemoci Duncana.

Klinické projevy X-vázanou lymfoproliferativní syndrom - onemocnění Duncan
X-vázaná lymfoproliferativní syndrom evidentní u chlapců pouze po infekci se Epstein-Barrové, obvykle do 5 let věku. Existují tři hlavní klinická fenotyp: 1) blesk, často fatální infekční mononukleóza (50%), 2), lymfom s výhodou B-buněk (25%) a 3) získané hypogamaglobulinémie (25%).
produkty protilátka na core antigenu Epstein-Barrové výrazně snížen, zatímco protilátky k kapsidový antigen viru může být dokonce mnohem vyšší, než je obvyklé. Prognóza je nepříznivá: 70% pacientů umírá chlapců do 10 let věku. Je známo, pouze 2 případy, kdy pacienti žijí déle než 40 let. Při absenci rodinnou anamnézou onemocnění diagnostikováno v dokud komplikace je obtížné stanovit, protože pacienti nedělají žádné stížnosti. Stejných rodinách ovlivněn chlapci nemoc může být diagnostikována na základě analýzy DNA před primárním viru.
Přibližně 50% z omezeného počtu pacienti, transplantační HLA-identický nefrakcionovaného kostní dřeně, se nadále žít a příznaky choroby nemají.
K dispozici jsou 2 rodiny, z nichž každá chlapci jeden řádek posloupnosti bylo odhaleno OVGGG, a na druhé straně - fulminantní infekční mononukleózu. Nicméně všichni pacienti v každé z linek sebou, navzdory různým klinického fenotypu, tam byl jeden a týž SH2D1A mutaci. Proto každý chlapec s diagnózou OVGGG, zvláště když existuje řada několika mužských pacientů by měla být podezřelá X-vázaná lymfoproliferativní syndrom.
 Hypotéza dva signály. Schéma interakce T a B lymfocytů
Hypotéza dva signály. Schéma interakce T a B lymfocytů Mechanismus aktivace lymfocytů klonů. Tvorba plazmatických buněk protilátek
Mechanismus aktivace lymfocytů klonů. Tvorba plazmatických buněk protilátek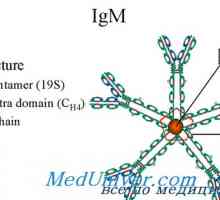 X-vázaná syndrom hyperproduction imunoglobulinových m (IgM) chlapců. Mutace CD40 CD154
X-vázaná syndrom hyperproduction imunoglobulinových m (IgM) chlapců. Mutace CD40 CD154 Celkové variabilní hypogamaglobulinémie (ovggg) u dětí. Příčina a kliniky
Celkové variabilní hypogamaglobulinémie (ovggg) u dětí. Příčina a kliniky Hypoplazii aplazie brzlíku. Syndrom di Giorgio
Hypoplazii aplazie brzlíku. Syndrom di Giorgio Autozomálně recesivní syndrom hyperproduction imunoglobulinu m (IgM). genová mutace pomoc
Autozomálně recesivní syndrom hyperproduction imunoglobulinu m (IgM). genová mutace pomoc Mechanismy primární imunitní odpovědi u plodu
Mechanismy primární imunitní odpovědi u plodu Porušení T-lymfocyty. CD8 lymfopenie
Porušení T-lymfocyty. CD8 lymfopenie Tvorba nk-fetální imunitní buňky. T-lymfocytů funkce imunity
Tvorba nk-fetální imunitní buňky. T-lymfocytů funkce imunity Komplikace transplantace ledvin. Důsledky imunosuprese
Komplikace transplantace ledvin. Důsledky imunosuprese Retikulární dysgeneze. purinová nukleosidová fosforyláza deficit
Retikulární dysgeneze. purinová nukleosidová fosforyláza deficit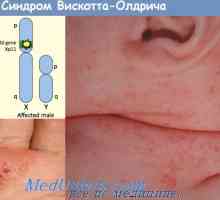 Wiskott-Aldrichův syndrom. Imunodeficience s trombocytopenií a ekzém
Wiskott-Aldrichův syndrom. Imunodeficience s trombocytopenií a ekzém Omenn syndrom. Immmunodefitsit při nedostatku MHC antigeny
Omenn syndrom. Immmunodefitsit při nedostatku MHC antigeny X-vázaná těžkou kombinovanou imunodeficiencí chlapců. Imunodeficience s deficitem adenosin deaminázy
X-vázaná těžkou kombinovanou imunodeficiencí chlapců. Imunodeficience s deficitem adenosin deaminázy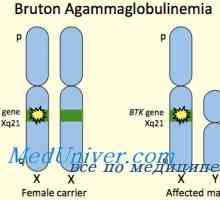 X-vázaná agamaglobulinémie. Brutonovskaya agamaglobulinémií děti
X-vázaná agamaglobulinémie. Brutonovskaya agamaglobulinémií děti T-lymfocyty. Charakteristika T-lymfocyty. Typy molekul na povrchu T-lymfocytů.
T-lymfocyty. Charakteristika T-lymfocyty. Typy molekul na povrchu T-lymfocytů.- Funkce v buňkách. Typy molekul na povrchu lymfocytů.
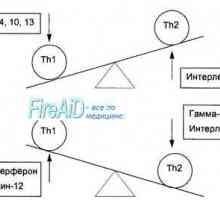 Populace T-lymfocytů. Subpopulace T-lymfocytů. CD4 T-lymfocyty. CD8 T-lymfocyty.
Populace T-lymfocytů. Subpopulace T-lymfocytů. CD4 T-lymfocyty. CD8 T-lymfocyty. B-lymfocyty. Charakterizace B-lymfocytů. paměťové buňky.
B-lymfocyty. Charakterizace B-lymfocytů. paměťové buňky.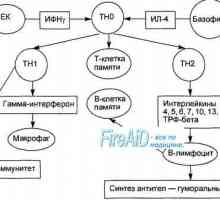 Aktivace T a B lymfocytů v imunitní odpovědi. Aktivace lymfocytů. Tvoří specifickou imunitní…
Aktivace T a B lymfocytů v imunitní odpovědi. Aktivace lymfocytů. Tvoří specifickou imunitní…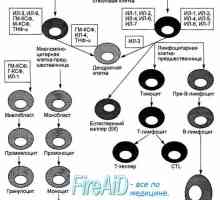 Tvoří specifickou imunitní odpověď. Imunologické paměti jako druh imunitní odpovědi.
Tvoří specifickou imunitní odpověď. Imunologické paměti jako druh imunitní odpovědi.