Anomálie ženských pohlavních orgánů. Syndromy Kaufman-mák-Cusick a Mayer-Rokitansky-Kuster-Hauser
Vrozené vady pohlavních orgánů nalezený v téměř 3% živě narozených dětí. Ty zahrnují abnormality vejcovodů, dělohy, děložního čípku a pochvy. Spektrum abnormalit včetně ageneze, atrézie a vzdělávací bariéry. U koncepce, žena embryo a diferenciace primárních gonad ve vaječníku degeneraci Wolfova vývodu potrubí a Mullerian diferencovat do reprodukčního systému u žen. Molekulární podstata vývoji samičího reprodukčního traktu člověka nejsou dobře pochopeny.
Víme, že mnohem více informací o tomto vývoj u myší díky experimenty s „knockout“ genů. Proces Müllerian konjugátu s apoptózou nebo programované buněčné smrti. Tento proces je velmi regulovaná, vyskytuje se zahrnující více genů, včetně bcl-2, chrání buňky před apoptózou. V nepřítomnosti tohoto genu ve správnou dobu, kdy byly buňky sjednoceni vedení připravený resorpce může nastat septum vytrvalost. Popsané lidské genetické syndromy, které mají vliv na procesy tvorby, diferenciace a involuce ženského reprodukčního trati.
K Genetické syndromy osoba, což vede k tvorbě patologii samičího reprodukčního ústrojí, jsou syndromy Kaufman-Mac Cusick a Mayer-Rokitansky-Kuster-Hauser, jakož i diabetes u mladých zralého typu, typ syndromu V. Kaufman-Mac Cusick - autosomální recesivní nemoc, původně popsané v Amish náboženská obec charakterizována polydaktylii, vrozených srdečních vad a gidrometrokolposom. Syndrom se může vyvinout u mužů, ale mají to zobrazeno pouze anomálie ruky a srdeční choroby.

nemožnost přeměny utero-vaginální deska kanál v průběhu embryogeneze vede k tvorbě příčnými přepážkami vaginalis, který způsobuje děložní natahování a stlačování přilehlých orgánů. Tyto přepážky mají tendenci být lokalizovány v horní třetině pochvy. Takový stav, plní se syndromem Bardet-Beadle spolu s retinopatie, poruchy učení, obezita, polydaktylii, renální abnormality a samčí hypogonadismus. MKKS gen kóduje protein, který funguje jako chaperonin - zástupce skupiny proteinů, které přispívají k optimalizaci prostorové struktury jiných proteinů po jejich uvolnění z ribozomy.
jak genové mutace MKKS způsobí rozvoj Kaufman syndrom-Mac Cusick nebo více z heterogenní syndrom, Bardet-Beadle, stále zůstává nejasný.
Syndromu Mayer-Rokitansky-Kuster-Hausera je také známý jako syndrom Rokitansky-Küster-Hauser. Tento syndrom Müllerian aplazie, ve kterém je zjištěna nedostatečná dělohy těla, čípku děložního a horní pochvy částmi. Ve většině případů pacientů - žen s karyotypem 46, XX, který zachránil vaječníky a normální zevní genitál. Také spojeny se syndromem renální patologie a někdy kostních abnormalit. Předpokládá se, že syndrom se dědí autosomálně recesivním způsobem, ale dosud nebyl identifikován příslušný gen.
léčba Jedná se o umělé vytvoření nového pochvy pomocí dilatátory. MURCS příznak zahrnuje Müllerian aplazie, ageneze ledvin, abnormality obratlů, Klippel-Feil syndrom a malým vzrůstem. Toto ojedinělé syndrom neznámé stále kauzální genetickou anomálii. Syndromy Fraser a Meckel-Gruber také spojován s Müllerian aplazií. Diabetes typu zralý mladý, typ V, - autozomálně dominantní patologie způsobená mutacemi v jaterních nukleární faktor (1b HNF-1b), a vyznačující se anomálií a renální aplazie Müllerian. Mutace v tomto genu může také způsobit Müllerian aplázie a v nepřítomnosti juvenilního diabetu.
Müllerian inhibiční faktor, také známý jako AMG - zástupce rodu transformačního růstového faktoru b (TGF-B), syntetizovaný podle pohlavních žláz. Po určení mužského gonad ve varlatech transformace umocňování Müllerian je jedním z prvních příznaků sexuální diferenciace mužského typu. V současné době dva typy popsané syndrom přetrvávající Mullerian - I a II. I typ I způsobena mutacemi AMG gen lokalizován na chromozómu 19. mutace v tomto genu detekován v 47% familiárních případů přetrvávající Müllerian syndromu, je přenášen v autosomálně recesivním způsobem.
Typ II způsobené mutací v genu receptor typu II, AMH, které vyvolávají 38% trvalé Müllerian syndromu. Obsah AMH séra těchto pacientů v normálním rozmezí. typ dědičnosti jako autozomálně recesivní. Pacienti se vyznačují karyotypem 46, XY, a jsou obvykle vytvořeny vnější mužské genitálie. Mohou rozvíjet kryptorchizmu spojené s tříselné kryptorchické gryzhami- často objevovat v průběhu chirurgického zákroku.

 Fraser syndrom. Diagnostika a léčení syndromu u plodu Fraser
Fraser syndrom. Diagnostika a léčení syndromu u plodu Fraser Systém mužského pohlavního kanálu embrya. Žena reprodukční systém plod kanály
Systém mužského pohlavního kanálu embrya. Žena reprodukční systém plod kanály Snížení vaječníky plodu. Základy pohlavních orgánů embrya
Snížení vaječníky plodu. Základy pohlavních orgánů embrya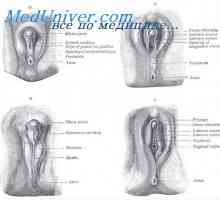 Anomálie dělohy a pochvy. fetální varle
Anomálie dělohy a pochvy. fetální varle Vývoj vnitřních pohlavních orgánů plodu. Záložka pohlavních žláz plodu
Vývoj vnitřních pohlavních orgánů plodu. Záložka pohlavních žláz plodu Mullerian vylučovací potrubí. vývoj plodu mužského vzoru
Mullerian vylučovací potrubí. vývoj plodu mužského vzoru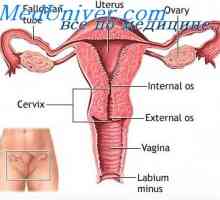 Fyziologie ženských pohlavních orgánů. Dámská hormonální systém
Fyziologie ženských pohlavních orgánů. Dámská hormonální systém Vrozené anomálie ledvin. ageneze ledvin
Vrozené anomálie ledvin. ageneze ledvin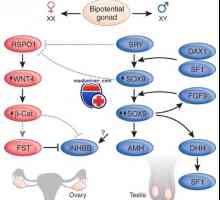 Mutace a genové duplikace dax1, sox9. pohlaví rozpor xy genotyp a kampomelicheskaya dysplazie
Mutace a genové duplikace dax1, sox9. pohlaví rozpor xy genotyp a kampomelicheskaya dysplazie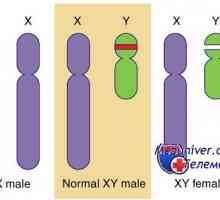 Genetické poruchy pohlavních žláz. Geny sry, WT1 a syndromy Fraser a Denis-dresha
Genetické poruchy pohlavních žláz. Geny sry, WT1 a syndromy Fraser a Denis-dresha Cysta-stop-genitální syndrom. Genetika mužské neplodnosti
Cysta-stop-genitální syndrom. Genetika mužské neplodnosti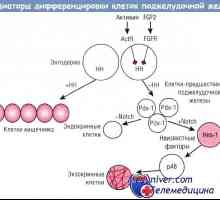 Regulace diferenciace exokrinních pankreatických buněk
Regulace diferenciace exokrinních pankreatických buněk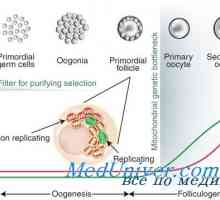 Nábor folikulů. Vliv na folikulů gonadotropinů
Nábor folikulů. Vliv na folikulů gonadotropinů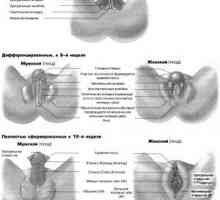 Vývoj pohlavních orgánů u plodu by týdny
Vývoj pohlavních orgánů u plodu by týdny Vrozené poruchy sexuální differentsirovkizabolevaniya způsobené chromozomálních abnormalit.…
Vrozené poruchy sexuální differentsirovkizabolevaniya způsobené chromozomálních abnormalit.…- Zdraví encyklopedie, nemoc, léky, lékař, lékárna, infekce, souhrny, sex, gynekologie, urologie.
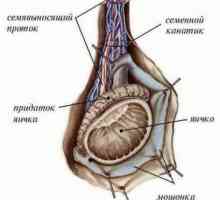 Diferenciace varlat a vaječníků
Diferenciace varlat a vaječníků Stanovení podlaha (spy jako určovací faktor varlete)
Stanovení podlaha (spy jako určovací faktor varlete) Müllerian anomálie
Müllerian anomálie Tuberkulóza ženských pohlavních orgánů, příznaky
Tuberkulóza ženských pohlavních orgánů, příznaky Vrozené vady pohlavních orgánů
Vrozené vady pohlavních orgánů