Retikulární dysgeneze. purinová nukleosidová fosforyláza deficit
reticular dysgeneze Poprvé byl popsán ve dvou chlapců, identických dvojčat, kteří byli zcela chybí lymfocytů a granulocytů v periferní krvi a kostní dřeni. Sedm z 8 dětí s touto vadou bylo zabito od 3 do 119 dnů od života nevyléčitelná infektsiy- sedm bylo zachráněno pomocí transplantace kostní dřeně. Thymus pacientů s hmotností nižší než 1 g, neexistovaly žádné Hassall je krvinky a sotva obsahoval thymocytů. Retikulární dysgeneze je považován za jednu SCID. Molekulární podstata autosomálně recesivní onemocnění není známa.
když kombinoval imunodeficience (PDC), na rozdíl od SCID funkci T-lymfocytů do určité míry zachována. Jako SCID, KID syndrom má různorodou genetický základ. U kojenců s výraznou KID opakovaným nebo chronickým plic, kůže a infekce močových cest, afty, chronický průjem a sepse způsobené Gram-negativními bakteriemi. Tyto děti jsou velmi obtížné tratě plané neštovice, které zaostávají ve vývoji. Ačkoli pacienti obecně dožívají vyššího věku než u SCID, ale stále umírají v raném dětství. Často definovat neutropenie a eosinofilie.
Obsah všech tříd imunoglobuliny v séru mohou být zvýšeny, ale někdy jsou izolovány deficit IgA, k výraznému zvýšení hladin IgE a zvýšená koncentrace IgD. Ve většině případů, poruchy produkce protilátek.

na Studie buněčné imunity nalezeno lymfopenie, ostrý deficit T-lymfocytů a výrazně oslabena proliferační odpověď lymfocytů mitogeny, antigeny a alogenních buněk in vitro. V paracortical látky periferní lymfatické tkáně je přítomen velmi málo buněk. Hodnota brzlíku a počet thymocytů prudce snizheny- Hassall je krvinky jsou obvykle chybí. Tento syndrom je dědičná, obvykle autozomálně recesivní.
purinová nukleosidová fosforyláza deficit
purinová nukleosidová fosforyláza deficit ve více než 40 pacientů s dítětem. Je založena na bodové mutace v genu enzymu se nachází na chromozomu 14. (ql3.1 části). V těchto případech je na rozdíl od nedostatku ADA, neexistují žádné porušení kostry, a hladiny kyseliny močové v krvi a moči je obvykle výrazně snížena. Příčinou smrti jsou vakcínie, plané neštovice, lymfosarkom nebo reakce „štěp versus hostitel“ zprostředkované allogenní T-buňky neozářené krve nebo kostní dřeně.
V 60-70% pacientů pozorované neurologické poruchy, 30% - autoimunitní onemocnění. Náhlé lymfopenie souvisí zejména s deficitem T-lymfocyty. Jejich úkolem je do jisté míry ovlivněn. Podíl krevních NK-buněk zvyšuje. Syndrom lze diagnostikovat prenatálně. Genová terapie - v budoucnosti, ale nyní jediný způsob léčby - transplantace kostní dřeně.
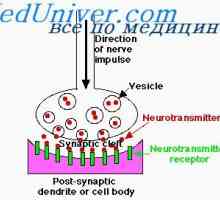 Příznaky vrozených imunodeficiencí. Nedostatek buněčné imunity u novorozence
Příznaky vrozených imunodeficiencí. Nedostatek buněčné imunity u novorozence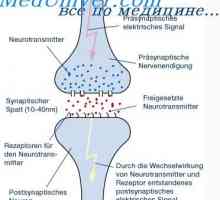 Těžkou kombinovanou imunodeficiencí. Morfologie brzlíku alymphoplasia
Těžkou kombinovanou imunodeficiencí. Morfologie brzlíku alymphoplasia Imunitní nedostatečnosti s achondroplazie. Morfologie imunodeficience s…
Imunitní nedostatečnosti s achondroplazie. Morfologie imunodeficience s… V kombinaci s imunodeficience fermentopathy. nezelofa syndrom nebo alymphocytosis
V kombinaci s imunodeficience fermentopathy. nezelofa syndrom nebo alymphocytosis Ataxie teleangiektázie u dětí. Imunodeficience s genovou mutací receptoru IFN
Ataxie teleangiektázie u dětí. Imunodeficience s genovou mutací receptoru IFN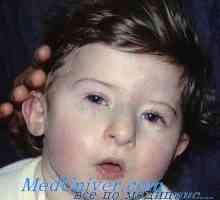 Hypoplazii aplazie brzlíku. Syndrom di Giorgio
Hypoplazii aplazie brzlíku. Syndrom di Giorgio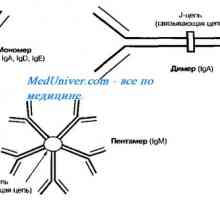 Autozomálně recesivní syndrom hyperproduction imunoglobulinu m (IgM). genová mutace pomoc
Autozomálně recesivní syndrom hyperproduction imunoglobulinu m (IgM). genová mutace pomoc Těžké kombinované imunodeficience u dětí. Příznaky a léčba
Těžké kombinované imunodeficience u dětí. Příznaky a léčba Immmunodefitsit s genu pro receptor mutace yl-2. Metafyzeální typ chondrodysplasia makové-Cusick
Immmunodefitsit s genu pro receptor mutace yl-2. Metafyzeální typ chondrodysplasia makové-Cusick X-vázaná lymfoproliferativní syndrom chlapců. Duncanova nemoc
X-vázaná lymfoproliferativní syndrom chlapců. Duncanova nemoc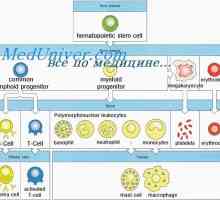 Tvorba imunity v plodu. novorozenecká lymfopoéze
Tvorba imunity v plodu. novorozenecká lymfopoéze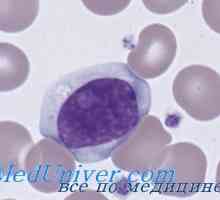 Imunodeficience při poruše JAK3 yl-7ra, RAG1 nebo rag2, CD45
Imunodeficience při poruše JAK3 yl-7ra, RAG1 nebo rag2, CD45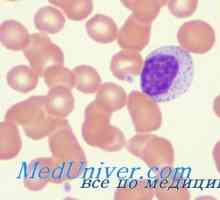 Omenn syndrom. Immmunodefitsit při nedostatku MHC antigeny
Omenn syndrom. Immmunodefitsit při nedostatku MHC antigeny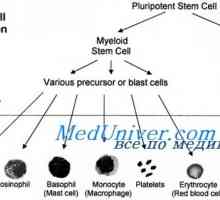 Transplantace kmenových buněk v srpkovitá anémie, syndrom Diamond Black fan
Transplantace kmenových buněk v srpkovitá anémie, syndrom Diamond Black fan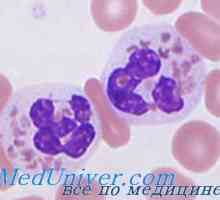 Dědičné neutropenie. Těžká kongenitální neutropenie syndrom Kostmann
Dědičné neutropenie. Těžká kongenitální neutropenie syndrom Kostmann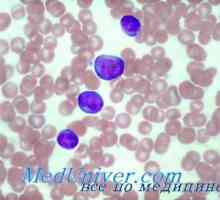 X-vázaná těžkou kombinovanou imunodeficiencí chlapců. Imunodeficience s deficitem adenosin deaminázy
X-vázaná těžkou kombinovanou imunodeficiencí chlapců. Imunodeficience s deficitem adenosin deaminázy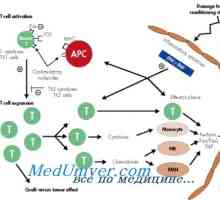 Chronické onemocnění štěpu proti hostiteli (GVHD)
Chronické onemocnění štěpu proti hostiteli (GVHD)- V Evropě se naučil zacházet s těžkou kombinovanou imunodeficiencí
- Infantilní vrozený glaukom. juvenilní glaukom
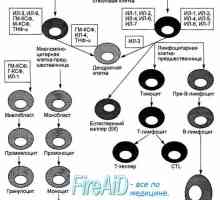 Kostní dřeně. funkce kostní dřeně. Mielomonotsitopoez.
Kostní dřeně. funkce kostní dřeně. Mielomonotsitopoez.- Timaktid (thymactidum). Komplexní polypeptidy z brzlíku (thymus) telat a jehňat. Indukuje…